Modelos de mutación y filogenética molecular. Bioinformática, Kevin Yip-CSE-CUHK (Universidad china de Hong-Kong)
|
|
|
- Gerardo Villalba Luna
- hace 6 años
- Vistas:
Transcripción
1 Modelos de mutación y filogenética molecular Bioinformática, Kevin Yip-CSE-CUHK (Universidad china de Hong-Kong)
2 HOY 1. Distancia evolutiva y modelos de mutación 2. Árboles: Las estructuras jerárquicas relacionando diferentes objetos biológicos 1. Formatos de archivo 2. reconstrucción de árboles filogenéticos 3. Métodos basados en secuencias máxima parsimonia Máxima verosimilitud 4. métodos basadosen distancia UPGMA Unión de vecinos
3 distancia evolutiva Supongamos que tenemos una alineación de dos secuencias. En una posición, una secuencia tiene A y otra tiene C. Supongamos que las secuencias tienen un ancestro común (por ejemplo son la misma proteína en ratones y ratas) Qué tenía el antepasado común en esa posición? No sabemos. Digamos A. Cuántas sustituciones han ocurrido desde su divergencia? Podría ser una (s 0 [3]:A s 2 [3]:C) Podría haber dos (por ejemplo, s 0 [3]:A s 2 ' [3]:G s 2 [3]:C) Podrían ser más un ejemplo con tres? Queremos una manera de definir la "distancia evolutiva" entre dos secuencias observadas de acuerdo con el número de mutaciones sucedido Cómo podemos definirla sin saber lo que ha sucedido? Necesitamos estimarla usando un modelo de mutaciones s 1 :TTAGG s 2 :TTCGG s 0 :TT? GG s 1 :TTAGG s 2 :TTCGG s 0 :TTAGG s 1 :TTAGG s 2 :TTCGG s 0 :TTAGG s 1 :TTAGG s 2 ' :TTGGG s 2 :TTCGG
4 modelo de mutación Un modelo de mutación es un modelo probabilístico que describe cómo ocurren las mutaciones a través del tiempo Con qué frecuencia ocurre una mutación? Qué tipo de mutaciones son más frecuentes? Para simplificar las cosas, vamos a hacer las siguientes suposiciones: Las posiciones son independientes Las tasas de mutación son las mismas para diferentes posiciones y en diferentes momentos de la historia Teniendo en cuenta el estado actual, los estados futuros no dependen de los estados pasados Sabemos que estos supuestos por lo general no son ciertos, pero sin ellos los cálculos pueden ser difíciles Existen modelos más complejos que requieren un menor número de supuestos fuertes. Hoy sólo estudiaremos modelos simples
5 El modelo Jukes Cantor Propuesto por Jukes y Cantor en 1969 Tasa de sustitución,, idéntica para las otras tres bases en una unidad de tiempo Suponemos que hay a lo sumo una mutación dentro de una unidad de tiempo Siempre podemos hacer que la unidad más Gpequeña de asegurar esto 1 3 U R 1 3 A G Nota: Una "sustitución" es N M una mutación puntual que O realmente ocurrió, mientras que una falta de coincidencia en una alineación podría ser causado por una o más C T sustituciones
6 Ilustración del modelo Jukes Cantor Supongamos que en el momento 0, la posición 1 de una secuencia era A En el momento 1: Hay una probabilidad de 1 3 de que la posición sea A Hay una probabilidad de de que la posición sea C Hay una probabilidad de de que la posición sea G Hay una probabilidad de de que la posición sea T En el momento 2, cuál es la probabilidad de que la posición sea A si sólo se sabe que era A en el tiempo 0, pero no se sabe lo que era en el momento 1? Dos posibilidades: 1. En el momento 1 era A, y no hubo ninguna mutación del instante 1 al 2 [probabilidad: (1 3 ) 2 ] 2. En el momento 1, era C,G ó T, y hubo una mutación a A del instante 1 al 2 [probabilidad: 3 2 ] Por lo tanto, la probabilidad total de que la posición sea A en el momento 2 es (1 3 )
7 fórmulas recursivas P X Y (t) es la probabilidad de que una base que era X en el instante 0, sea Y en el instante t P A A (1) = 1 3 P A A (2) = (1 3 ) En general, P A A (t+1) = (1 3 )P A A (t) + [1 P A A (t)] En general: P X X (t+1) = (1 3 )P X X (t) + [1 P X X (t)] P X Y (t+1) = [1 P X X (t+1)] / 3 para cualquier X para cualquier X, Y En primer lugar, estudiamos cómo calcular P X Y (t) para un ratio de mutación y tiempo de divergencia t, después estudiamos cómo podemos utilizar P X Y (t) para estimar el número de mutaciones que han ocurrido desde la divergencia de las dos secuencias
8 Solución de P A A (t) P A A (t+1) = (1 3 )P A A (t) + [1 P A A (t)] P A A (t) P A A (t+1) P A A (t) = (1 3 )P A A (t) + [1 P A A (t)] P A A (t) = [1 4P A A (t)] Para una unidad de tiempo infinitamente pequeña, se obtiene una ecuación diferencial de primer orden, que puede ser resuelta mediante el uso de un factor de integración dp A A d α 1 4P A A P A A 1 4 P A A e 4α e 4α Observación: Cuando t es grande, el estado inicial (es decir, nucleótido) ya no importa y las cuatro bases son igualmente probables
9 fórmulas finales para P X Y (t) Por simetría, P A A P C C P G G P T T e 4α Del mismo modo, es fácil demostrar que P A C P A G P A T P T G e 4α e 4α
10 Volviendo al problema Qué hemos hecho hasta ahora? Dado 1. El estado ancestral de un posición 2. La tasa de sustitución (probabilidad de cada tipo de mutación en una unidad de tiempo) Determinar la probabilidad del estado actual, que es t unidades de tiempo después del evento de separación, donde t también se da Qué es lo que realmente queremos? Teniendo en cuenta el estado actual de dos secuencias El estado ancestral se desconoce Determinar el número de sustituciones que sucedió en las dos secuencias desde su divergencia, tantoobservadascomono observadas La tasa de sustitución ( ) y hace cuánto tiempo que las dos secuencias han divergido (t) son también desconocidos
11 Las dificultades y las ideas Dificultad # 1: No sabemos el estado ancestral, tasa de mutación ó tiempo de divergencia t soluciones: Debido a la simetría, el estado ancestral No importa si sólo nos preocupa si dos secuencias actuales tienen el mismo nucleótido o no en cada posición P mismo (t) = [P A A (t)] 2 + [P A C (t)] 2 + [P A G (t)] 2 + [P A T (t)] 2 = [P C A (t)] 2 + [P C C (t)] 2 + [P C G (t)] 2 + [P C T (t)] 2 = [P G A (t)] 2 + [P G C (t)] 2 + [P G G (t)] 2 + [P G T (t)] 2 = [P T A (t)] 2 + [P T C (t)] 2 + [P T G (t)] 2 + [P T T (t)] 2 No conocemos ó t, pero se puede estimar fácilmente su producto t y resulta que esto es todo lo que necesitamos
12 Las dificultades y las ideas Dificultad # 2: Aunque conociéramos el estado ancestral, la tasa de mutación y el tiempo de divergencia t, todavía habría un número infinito de posibilidades, cada una con una cierta probabilidad Solución: Vamos a hablar del número esperado de mutaciones ocurridas, es decir, el promedio de todos los casos considerando el número de mutaciones de cada uno y la probabilidad de que ocurra
13 Cómo ayudan esperanza y varianza en la solución de nuestro problema? No sabemos si una mutación aparece después de una unidad de tiempo, pero si tenemos en cuenta un gran número de unidades de tiempo, el número esperado de mutaciones por unidad de tiempo es 3 t Si podemos estimar el número de mutaciones que han sucedido, podemos calcular 3 t aunque nunca sabemos los valores de y t por separado Por eso utilizamos la fórmula de aproximación para P A A (t) = 3/4 e 4 t + 1/4 sólo usa el producto t
14 Estimando el número de sustituciones Para el modelo de Jukes Cantor: Para una sola posición, de probabilidad para dos secuencias separa t unidades de hace tiempo que tiene el mismo estado es (suponiendo que el estado ancestral era UN, Pero la misma fórmula es válida para otros estados ancestrales): Idea # 1 P A A 2 P A C 2 P A G 2 P A T e 4α e 4α e 4α 9 16 e 8α e 4α 3 16 e 8α e 8α
15 Estimando el número de sustituciones Para el modelo de Jukes Cantor: En consecuencia, la probabilidad de que las dos secuencias tengan diferentes estados en una posición es Idea # 2 p diff e 8α e 8α α 1 8 ln p diff No sabemos el valor de (tasa de sustitución) o t (tiempo desde la divergencia de las dos secuencias), pero podemos estimar p diff, lo que nos dará una estimación de t.
16 Estimando el número de sustituciones Cómo estimar p diff, la probabilidad de que dos secuencias aleatorias generadas según el procedimiento anterior tengan diferentes estados en una posición? Estimamos p diff por x/n, Donde x es el número de posiciones diferentes entre las secuencias observadas nuestra mejor suposición basada en datos observados Poniendo todo junto: Supongamos que tenemos dos secuencias de longitud n que divergieron hace tiempo t con x diferencias Sea K sup el no. de sustituciones por posición ocurridas para las dos secuencias desde su divergencia Según el modelo de Jukes Cantor, el valor esperado de K sup es (de la página anterior) E K sup 2 3α ln p diff 3 4 ln p diff 3 ln para n grande, la varianza de esta estimación es de aproximadamente p diff p diff p diff / /
17 Estimando el número de sustituciones Vamos a ver cómo podemos aplicar los resultados. Ejemplo: Supongamos que dos secuencias, cada una con n= 200 nucleótidos tienenx= 66 diferencias observadas, luego p diff = x/n = 66/200 = 0,33 E K sup 3 4 ln p diff 0.43 La varianza de esta estimación es p diff p diff p diff observaciones: 1. El número observado de sustituciones por posición es menor que el número estimado de sustituciones (observadas + no observadas) por posición, como se esperaba 2. La variación es bastante grande el número real puede ser un poco diferente de esta estimación (sería más pequeño para los n grandes)
18 Comprobación con nuestros datos de simulación Suponer = 0,005, t= 50 y n= 100 Dos copias de la misma secuencia aleatoria se generaron en t= 0, seguido de mutaciones independientes de acuerdo con el modelo de Jukes Cantor Resultados de la simulación de 10 ensayos: Trial x pdiff E[Ksup] (from formula) Variance of E[Ksup] estimation (from formula) Actual number of substitutions per site Actual variance of 10 trials 0.03
19 distancia evolutiva Ahora, podemos usar E [K sup ] como una medida de la distancia evolutiva entre dos secuencias alineadas sin indeles. Utilizar un modelo de mutación para definir la distancia es más robusto que la definición de la matriz de puntuación de sustitución arbitraria (como coincidencia = 1, diferencia = 1). Por supuesto, tenemos que alinear las secuencias en primer lugar, momento en el que todavía necesitamos una matriz de sustitución para empezar Hay otros modelos que permiten Más parámetros (por ejemplo, diferentes subtipos de sustituciones) Tasas variables en diferentes posiciones Dependencia entre diferentes posiciones Cambio de las tasas de sustitución con el tiempo indeles
20 HOY 1. Distancia evolutiva y modelos de mutación 2. Árboles: Las estructuras jerárquicas relacionando diferentes objetos biológicos 1. Formatos de archivo 2. reconstrucción de árboles filogenéticos 3. Métodos basados en secuencias máxima parsimonia Máxima verosimilitud 4. métodos basadosen distancia UPGMA Unión de vecinos

21 Clasificación de las especies Dominios y reinos Crédito de la imagen: Wikipedia, ed/classif gifs/animal class example.gif El Reino animal


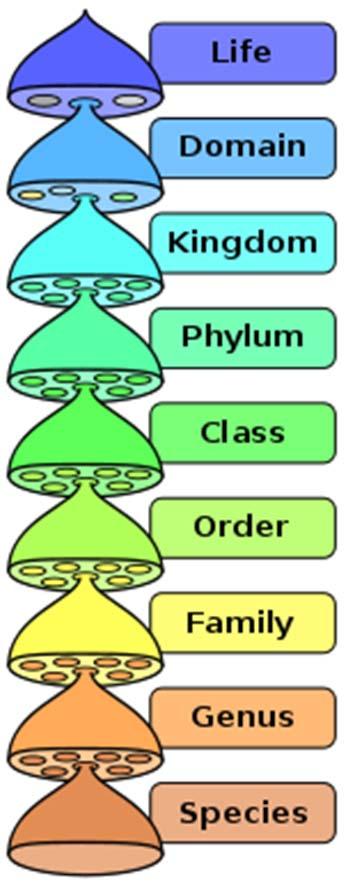


22 taxonomía Crédito de la imagen: Wikipedia,
, http://www.accessexcellence.org/rc/vl/gg/images/pedigree.gif, http://www.")
23 escalas más finas La misma idea se puede aplicar a clasificar diferentes cepas de un tipo de bacteria O incluso las relaciones familiares Crédito de la imagen: Hershberg et al, Genome Biology. 8: R164 (2007),

24 Relacionar objetos biológicos Cómo se determinaron las jerarquías? Especie: tradicionalmente por las similitudes morfológicas y de comportamiento, o evidencias paleontológicas Las cepas bacterianas: por propiedades biológicas físicas, químicas y Pregunta: Qué características debe ser usadas antes? Miembros de la familia: por pedigrí / genealogía Crédito de la imagen:
25 Filogenia Una manera sistemática y objetiva para construir estos árboles es mediante la comparación de las secuencias de DNA / proteínas Estudiamos aquí los árboles que relacionan los objetos suficientemente diferentes Especies diferentes Las diferentes cepas / poblaciones de una especie Nuestro objetivo es reconstruir las relaciones evolutivas reales en base a secuencias observables
26 suposiciones supuestos básicos detrás de los árboles filogenéticos: 1. Las secuencias actuales comparten un ancestro común 2. Todas mutaron a partir del ancestro común 3. Las mutaciones son raras. Por tanto, si los DNA de A y B son más similares que los de A y C, y que los de B y C, probablemente C se separó de A y B antes de su separación ancestro común de A, B y C ancestro común de A y B tiempo A B C

27 longitud de la rama Terminología Un árbol es un grafo acíclico con nodos conectados por aristas Un árbol filogenético es un árbol binario con secuencias (nodos) conectados por ramas (aristas) Los nodos hoja son las secuencias observadas Los nodos internos son las secuencias ancestrales no observadas El nodo raíz es el ancestro común de todas las secuencias observadas Longitudes de las ramas pueden representar distancias evolutivas Raíz Un nodo interno Una rama Un nodo hoja Crédito de la imagen: Hershberg et al, Genome Biology. 8: R164 (2007),
28 Árboles con y sin raíz A veces no está muy claro donde incluir el ancestro común Podemos tener un árbol sin raíz Crédito de la imagen: Lowery et al,. oncogén 24 (2): , (2005)
29 HOY 1. Distancia evolutiva y modelos de mutación 2. Árboles: Las estructuras jerárquicas relacionando diferentes objetos biológicos 1. Formatos de archivo 2. reconstrucción de árboles filogenéticos 3. Métodos basados en secuencias máxima parsimonia Máxima verosimilitud 4. métodos basadosen distancia UPGMA Unión de vecinos
30 formatos de archivo comunes para los árboles filogenéticos Newick (paréntesis anidados, con distancias) NEXUS (dando identificaciones cortas para secuencias, con más metadatos) PhyloXML (utilizando la estructura de XML)
31 formatos de archivo comunes Ejemplo newick: ((((Homo: 0,21, Pongo: 0,21): 0,28, Macaca: 0,49): 0,13, Ateles: 0,62): 0,38, Galago: 1,00); Representación grafica: NEXUS: END; Crédito de la imagen: BEGIN TAXA; DIMENSIONS NTAX = 5; TAXLABELS Homo Pongo Macaca Ateles Galago ; END; BEGIN TREES; TRANSLATE 1 Homo, 2 Pongo, 3 Macaca, 4 Ateles, 5 Galago ; TREE * UNTITLED = [&R] ((((1:0.21,2:0.21):0.28,3:0.49):0.13,4:0.62):0.38,5:1 );
32 formatos de archivo comunes PhyloXML <phyloxml xmlns:xsi=" xmlns=" xsi:schemalocation=" <phylogeny rooted="true"> <name>alcohol dehydrogenases</name> <description>contains examples of commonly used elements</description> <clade> <events> <speciations>1</speciations> </events> <clade> <taxonomy> <id provider="ncbi">6645</id> <scientific_name>octopus vulgaris</scientific_name> </taxonomy> <sequence> <accession source="uniprotkb">p81431</accession> <name>alcohol dehydrogenase class-3</name> </sequence> </clade>... </clade> </phylogeny> </phyloxml> Fuente de información:
33 El formato Newick Utiliza corchetes y coma para agrupar dos subárboles Usa dos puntos para indicar la distancia a los padres, si está disponible Termina con un punto y coma Representación grafica: newick: Crédito de la imagen: ((((Homo: 0,21, Pongo: 0,21): 0,28, Macaca: 0,49): 0,13, Ateles: 0,62): 0,38, Galago: 1,00); observaciones: Para un árbol sin raíz, de una manera sencilla para representarlo utilizando el formato Newick es poniendo una raíz de manera arbitraria Se pueden nombrar a los nodos internos, dando la etiqueta después del corchete de cierre (por ejemplo, (Homo: 0,21, Pongo: 0,21) HP: 0,28
34 HOY 1. Distancia evolutiva y modelos de mutación 2. Árboles: Las estructuras jerárquicas relacionando diferentes objetos biológicos 1. Formatos de archivo 2. reconstrucción de árboles filogenéticos 3. Métodos basados en secuencias máxima parsimonia Máxima verosimilitud 4. métodos basadosen distancia UPGMA Unión de vecinos
35 reconstrucción de árboles filogenéticos problema general: Dado un conjunto de secuencias / proteínas de DNA, encontrar un árbol filogenético de tal manera que probablemente corresponde a los eventos evolutivos históricos reales, describiendo: Cómo se conectan los nodos: Orden de eventos de separación Lo secuencias en los nodos internos: secuencias ancestrales Cuánto tiempo ha pasado desde que la separación: longitud de rama Hay varias formas de evaluar la probabilidad de que un árbol sea correcto. Vamos a estudiarlas "Re" Construcción: El árbol fue definido por la historia. Nosotros sólo tratamos de reconstruirlo desde las secuencias observadas
36 Qué secuencia usar? Si estamos estudiando un gen secuencia de DNA / proteína del gen Si queremos conocer la relación entre las diferentes especies el genoma completo (puede no ser factible) Algunos genes que evolucionan lentamente RNA ribosomal
37 La complejidad del problema Encontrar el "mejor" árbol es un problema difícil Cuántas (es decir, ignorando longitudes de rama y el orden de izquierda a derecha) son las topologías de árboles para un conjunto de k secuencias? Para los árboles enraizados: k = 2: 1 posibile topología de árbol k = 3: 3 ramas posibles para añadir # 3 k = 4: 5 posibles para añadir # 4, y así sucesivamente Por lo tanto el número de topologías de árboles es (2k 3) Exponencial Una vez más, la enumeración de todas las topologías para encontrar la mejor opción es imposible k Num. of rooted tree topologies , , ,027, ,459, ,729, ,749,310, ,234,143, ,905,853,580, ,458,046,676, ,190,283,353,629, ,898,783,962,511, ,332,659,870,762,850, ,643,095,476,700,000, ,200,794,532,637,890,000,000
38 La complejidad de un problema k Num. of rooted tree topologies Num. of unrooted tree topologies Del mismo modo, para los árboles sin raíces, k = 2: 1 topología k = 3: 1 forma de añadir # 3 k = 4: 3 formas de agregar # 4 k = 5: 5 formas de añadir # 5 El número de topologías es (2k 5) , ,135 10, ,027, , ,459,425 2,027, ,729,075 34,459, ,749,310, ,729, ,234,143,225 13,749,310, ,905,853,580, ,234,143, ,458,046,676,875 7,905,853,580, ,190,283,353,629, ,458,046,676, ,898,783,962,511,000 6,190,283,353,629, ,332,659,870,762,850, ,898,783,962,511, ,643,095,476,700,000,000 6,332,659,870,762,850, ,200,794,532,637,890,000, ,643,095,476,700,000,
39 La solución del problema: Ideas Qué se hace cuando se encuentra un problema difícil computacionalmente? Definir una versión más fácil del problema hacer ciertos supuestos Diseñar algoritmos / estructuras de datos inteligentes para evitar cálculos redundantes Usar heurísticas para resolverlo, no obteniendo necesariamente la solución óptima
40 métodos de reconstrucción de árboles filogenéticos Existen dos tipos principales de métodos: basados en secuencias: necesitará las secuencias métodos de parsimonia (problemas más fáciles, algoritmos inteligentes) métodos probabilísticos (problemas más fáciles, algoritmos inteligentes) Máxima verosimilitud bayesiano... basados en la distancia: solo dependen de las distancias entre las secuencias UPGMA (heurística) vecino más próximo (heurística)... Vamos a estudiar algunos de estos algoritmos
41 El próximo día 1. Distancia evolutiva y modelos de mutación 2. Árboles: Las estructuras jerárquicas relacionando diferentes objetos biológicos 1. Formatos de archivo 2. reconstrucción de árboles filogenéticos 3. Métodos basados en secuencias máxima parsimonia Máxima verosimilitud 4. métodos basadosen distancia UPGMA Unión de vecinos
TÉCNICAS INTELIGENTES EN BIOINFORMÁTICA. Evolución Molecular y Filogenia
 TÉCNICAS INTELIGENTES EN BIOINFORMÁTICA Evolución Molecular y Filogenia Ignacio Pérez Hurtado de Mendoza Grupo de investigación en Computación Natural Dpto. Ciencias de la Computación e Inteligencia Artificial
TÉCNICAS INTELIGENTES EN BIOINFORMÁTICA Evolución Molecular y Filogenia Ignacio Pérez Hurtado de Mendoza Grupo de investigación en Computación Natural Dpto. Ciencias de la Computación e Inteligencia Artificial
Conceptos básicos de filogenética molecular
 Dr. Eduardo A. RODRÍGUEZ TELLO CINVESTAV-Tamaulipas 18 de julio del 2013 Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Conceptos básicos de filogenética molecular 18 de julio del 2013 1 / 43 1 Conceptos básicos
Dr. Eduardo A. RODRÍGUEZ TELLO CINVESTAV-Tamaulipas 18 de julio del 2013 Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Conceptos básicos de filogenética molecular 18 de julio del 2013 1 / 43 1 Conceptos básicos
Construcción y análisis de árboles filogenéticos. Antonio Gómez Tato
 Construcción y análisis de árboles filogenéticos Antonio Gómez Tato Introducción Árboles filogenéticos Surgen a partir de la teoría de la evolución de Darwin. Son representaciones gráficas de las relaciones
Construcción y análisis de árboles filogenéticos Antonio Gómez Tato Introducción Árboles filogenéticos Surgen a partir de la teoría de la evolución de Darwin. Son representaciones gráficas de las relaciones
Árboles Filogenéticos. BT7412, CC5702 Bioinformática Diego Arroyuelo. 2 de noviembre de 2010
 Unidad 6: Árboles Filogenéticos BT7412, CC5702 Bioinformática Diego Arroyuelo 2 de noviembre de 2010 Temario (Introduction to Computational Molecular Biology Setubal y Meidanis Capítulo 6) 1. Introducción
Unidad 6: Árboles Filogenéticos BT7412, CC5702 Bioinformática Diego Arroyuelo 2 de noviembre de 2010 Temario (Introduction to Computational Molecular Biology Setubal y Meidanis Capítulo 6) 1. Introducción
Filogenias. Charles Darwin (1859)
") Filogenias Charles Darwin (1859) Filogenia Consiste en el estudio de las relaciones evolutivas entre diferentes grupos de organismos. Su inferencia implica el desarrollo de hipótesis sobre los patrones
Filogenias Charles Darwin (1859) Filogenia Consiste en el estudio de las relaciones evolutivas entre diferentes grupos de organismos. Su inferencia implica el desarrollo de hipótesis sobre los patrones
Análisis y Complejidad de Algoritmos. Arboles Binarios. Arturo Díaz Pérez
 Análisis y Complejidad de Algoritmos Arboles Binarios Arturo Díaz Pérez Arboles Definiciones Recorridos Arboles Binarios Profundidad y Número de Nodos Arboles-1 Arbol Un árbol es una colección de elementos,
Análisis y Complejidad de Algoritmos Arboles Binarios Arturo Díaz Pérez Arboles Definiciones Recorridos Arboles Binarios Profundidad y Número de Nodos Arboles-1 Arbol Un árbol es una colección de elementos,
Árboles. Un grafo no dirigido es un árbol si y sólo si existe una ruta unica simple entre cualquiera dos de sus vértices.
 ÁRBOLES Árboles Un grafo conectado que no contiene circuitos simples. Utilizados desde 1857, por el matemático Ingles Arthur Cayley para contar ciertos tipos de componentes químicos. Un árbol es un grafo
ÁRBOLES Árboles Un grafo conectado que no contiene circuitos simples. Utilizados desde 1857, por el matemático Ingles Arthur Cayley para contar ciertos tipos de componentes químicos. Un árbol es un grafo
Elvira Mayordomo y Jorge Álvarez. Marzo - Abril de 2016
 TRABAJO DE PRÁCTICAS Elvira Mayordomo y Jorge Álvarez Marzo - Abril de 2016 1 Introducción El trabajo de prácticas de la asignatura consistirá en que cada alumno realice por separado el trabajo que se
TRABAJO DE PRÁCTICAS Elvira Mayordomo y Jorge Álvarez Marzo - Abril de 2016 1 Introducción El trabajo de prácticas de la asignatura consistirá en que cada alumno realice por separado el trabajo que se
Filogenias. Inferencia filogenética
 Filogenias Para Darwin la evolución es descendencia con modificación a partir de un único origen de la vida. Siguiendo esta idea, todos los taxa actuales tendrán algún tipo de parentesco más o menos cercano.
Filogenias Para Darwin la evolución es descendencia con modificación a partir de un único origen de la vida. Siguiendo esta idea, todos los taxa actuales tendrán algún tipo de parentesco más o menos cercano.
MAXIMA PARSIMONIA EN LA INFERENCIA FILOGENÉTICA DE SECUENCIAS DE ADN
 MAXIMA PARSIMONIA EN LA INFERENCIA FILOGENÉTICA DE SECUENCIAS DE ADN - Inferir una filogenia es un proceso de estimación. Se hace la mejor estimación de una historia evolutiva con base en la información
MAXIMA PARSIMONIA EN LA INFERENCIA FILOGENÉTICA DE SECUENCIAS DE ADN - Inferir una filogenia es un proceso de estimación. Se hace la mejor estimación de una historia evolutiva con base en la información
Clase 2. Introducción a la Sistemática Cladística
 Curso: Principios y métodos de la Sistemática Cladística. Clase 2. Introducción a la Sistemática Cladística Fernando Pérez-Miles Entomología, Facultad de Ciencias El método más utilizado Hennig 950 Grundrüge
Curso: Principios y métodos de la Sistemática Cladística. Clase 2. Introducción a la Sistemática Cladística Fernando Pérez-Miles Entomología, Facultad de Ciencias El método más utilizado Hennig 950 Grundrüge
Lección 4. Métodos filogenéticos
 Básico La inferencia filogenética es un campo per se del estudio de la evolución, en continuo movimiento y expansión. filogenéticos La inferencia filogenética es un procedimiento de estimación estadística.
Básico La inferencia filogenética es un campo per se del estudio de la evolución, en continuo movimiento y expansión. filogenéticos La inferencia filogenética es un procedimiento de estimación estadística.
La teoría de coalescencia
 La teoría de coalescencia Proceso estocástico Es un linaje de alelos proyectado hacia el pasado (su ancestro común mas reciente) Aproximación básica usando el modelo de Wright-Fisher Modelo probabilístico
La teoría de coalescencia Proceso estocástico Es un linaje de alelos proyectado hacia el pasado (su ancestro común mas reciente) Aproximación básica usando el modelo de Wright-Fisher Modelo probabilístico
Código de barras del ADN. Dra. Analía A. Lanteri División Entomología- Museo de La Plata
 Código de barras del ADN Dra. Analía A. Lanteri División Entomología- Museo de La Plata CÓDIGO DE BARRAS DEL ADN Los genes están formados por EXONES (traducen a proteínas) y los INTRONES (no codificantes)
Código de barras del ADN Dra. Analía A. Lanteri División Entomología- Museo de La Plata CÓDIGO DE BARRAS DEL ADN Los genes están formados por EXONES (traducen a proteínas) y los INTRONES (no codificantes)
ÁRBOLES FILOGENÉTICOS
 ÁRBOLES FILOGENÉTICOS Por qué usar filogenias? El conocimiento del pasado es importante para poder resolver muchas cuestiones relacionadas con procesos biológicos. Las filogenias nos permiten obtener relaciones
ÁRBOLES FILOGENÉTICOS Por qué usar filogenias? El conocimiento del pasado es importante para poder resolver muchas cuestiones relacionadas con procesos biológicos. Las filogenias nos permiten obtener relaciones
MATRIZ DE ARBOLES DE DECISION
 MATRIZ DE ARBOLES DE DECISION Los árboles son un subconjunto importante de los grafos, y son una herramienta útil para describir estructuras que presentan algún tipo de jerarquía. Las dificultades de las
MATRIZ DE ARBOLES DE DECISION Los árboles son un subconjunto importante de los grafos, y son una herramienta útil para describir estructuras que presentan algún tipo de jerarquía. Las dificultades de las
Alineamiento múltiple de secuencias
 Dr. Eduardo A. RODRÍGUEZ TELLO CINVESTAV-Tamaulipas 11 de junio del 2013 Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Alineamiento múltiple de secuencias 11 de junio del 2013 1 / 39 1 Alineamiento múltiple de
Dr. Eduardo A. RODRÍGUEZ TELLO CINVESTAV-Tamaulipas 11 de junio del 2013 Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Alineamiento múltiple de secuencias 11 de junio del 2013 1 / 39 1 Alineamiento múltiple de
Práctica 1: Alineamientos
 Práctica 1: Alineamientos Partimos de un archivo de datos que contiene 5 secuencias de mrna asociado a la CFTR http://madma.usc.es/cursoverano/materiales Abrimos el ClustalX en la carpeta C:\ClustalX de
Práctica 1: Alineamientos Partimos de un archivo de datos que contiene 5 secuencias de mrna asociado a la CFTR http://madma.usc.es/cursoverano/materiales Abrimos el ClustalX en la carpeta C:\ClustalX de
Introducción a Árboles Árboles Binarios
 Introducción a Árboles Árboles Binarios Estructuras de Datos Andrea Rueda Pontificia Universidad Javeriana Departamento de Ingeniería de Sistemas Introducción a Árboles Estructuras hasta ahora Estructuras
Introducción a Árboles Árboles Binarios Estructuras de Datos Andrea Rueda Pontificia Universidad Javeriana Departamento de Ingeniería de Sistemas Introducción a Árboles Estructuras hasta ahora Estructuras
Lección 3. Modelos de evolución molecular. Transiciones. Transversiones. Transiciones. Inferencia filogenética Mutación y substitución
 Mutación y substitución Mutación (µ: cambio de base en la secuencia de DN de evolución molecular Substitución (µ: mutación que se fija en una población (o especie P(substitución = Nºmutaciones * P(fijación
Mutación y substitución Mutación (µ: cambio de base en la secuencia de DN de evolución molecular Substitución (µ: mutación que se fija en una población (o especie P(substitución = Nºmutaciones * P(fijación
Teoría de probabilidades
 Modelos Probabilistas Teoría de probabilidades Teoría de probabilidades Definiremos como probabilidad a priori (P(a)) asociada a una proposición como el grado de creencia en ella a falta de otra información
Modelos Probabilistas Teoría de probabilidades Teoría de probabilidades Definiremos como probabilidad a priori (P(a)) asociada a una proposición como el grado de creencia en ella a falta de otra información
Caracteres moleculares
 PEDECIBA BIOLOGÍA - 2007 Sistemática Biológica: Métodos y Principios Alejandro D Anatro Caracteres moleculares Esquema para esta clase Generalidades Tipos de caracteres moleculares Particularidades del
PEDECIBA BIOLOGÍA - 2007 Sistemática Biológica: Métodos y Principios Alejandro D Anatro Caracteres moleculares Esquema para esta clase Generalidades Tipos de caracteres moleculares Particularidades del
Curso de Inteligencia Artificial
 Curso de Inteligencia Artificial Modelos Ocultos de Markov Gibran Fuentes Pineda IIMAS, UNAM Redes Bayesianas Representación gráfica de relaciones probabilísticas Relaciones causales entre variables aleatorias
Curso de Inteligencia Artificial Modelos Ocultos de Markov Gibran Fuentes Pineda IIMAS, UNAM Redes Bayesianas Representación gráfica de relaciones probabilísticas Relaciones causales entre variables aleatorias
ÁRBOLES CRISTIAN ALFREDO MUÑOZ ÁLVAREZ JUAN DAVID LONDOÑO CASTRO JUAN PABLO CHACÓN PEÑA EDUARDO GONZALES
 ÁRBOLES CRISTIAN ALFREDO MUÑOZ ÁLVAREZ JUAN DAVID LONDOÑO CASTRO JUAN PABLO CHACÓN PEÑA EDUARDO GONZALES ÁRBOL Un árbol es un grafo no dirigido, conexo, sin ciclos (acíclico), y que no contiene aristas
ÁRBOLES CRISTIAN ALFREDO MUÑOZ ÁLVAREZ JUAN DAVID LONDOÑO CASTRO JUAN PABLO CHACÓN PEÑA EDUARDO GONZALES ÁRBOL Un árbol es un grafo no dirigido, conexo, sin ciclos (acíclico), y que no contiene aristas
Fundamentos y Aplicaciones Prácticas del Descubrimiento de Conocimiento en Bases de Datos. - Sesión 11 -
 Fundamentos y Aplicaciones Prácticas del Descubrimiento de Conocimiento en Bases de Datos - Sesión 11 - Juan Alfonso Lara Torralbo 1 Índice de contenidos Actividad. Resolución manual de clasificación bayesiana
Fundamentos y Aplicaciones Prácticas del Descubrimiento de Conocimiento en Bases de Datos - Sesión 11 - Juan Alfonso Lara Torralbo 1 Índice de contenidos Actividad. Resolución manual de clasificación bayesiana
Un árbol binario T se define como un conjunto finito de elementos, llamados nodos, de forma que:
 Instituto Universitario de Tecnología Industrial Rodolfo Loero Arismendi I.U.T.I.R.L.A. ÁRBOLES Sección 3DA Asignatura: Estructura de Datos Lenguaje (C). Ciudad Bolívar _ abril_ 2006. Introducción El siguiente
Instituto Universitario de Tecnología Industrial Rodolfo Loero Arismendi I.U.T.I.R.L.A. ÁRBOLES Sección 3DA Asignatura: Estructura de Datos Lenguaje (C). Ciudad Bolívar _ abril_ 2006. Introducción El siguiente
Teorema Central del Límite (1)
") Teorema Central del Límite (1) Definición. Cualquier cantidad calculada a partir de las observaciones de una muestra se llama estadístico. La distribución de los valores que puede tomar un estadístico
Teorema Central del Límite (1) Definición. Cualquier cantidad calculada a partir de las observaciones de una muestra se llama estadístico. La distribución de los valores que puede tomar un estadístico
Consenso: Hoy 3 temas. 2- evaluando los resultados. evolucion.fcien.edu.uy/sistematica/sistematica.htm
 PEDECIBA BIOLOGÍA CURSO: SISTEMÁTICA BIOLÓGICA: MÉTODOS Y PRINCIPIOS Guillermo D Elía Hoy 3 temas 1- qué hacemos cuando al analizar una matriz obtenemos más de un árbol más óptimo? - consensos - pesos
PEDECIBA BIOLOGÍA CURSO: SISTEMÁTICA BIOLÓGICA: MÉTODOS Y PRINCIPIOS Guillermo D Elía Hoy 3 temas 1- qué hacemos cuando al analizar una matriz obtenemos más de un árbol más óptimo? - consensos - pesos
Estimación de Parámetros. Jhon Jairo Padilla A., PhD.
 Estimación de Parámetros Jhon Jairo Padilla A., PhD. Inferencia Estadística La inferencia estadística puede dividirse en dos áreas principales: Estimación de Parámetros Prueba de Hipótesis Estimación de
Estimación de Parámetros Jhon Jairo Padilla A., PhD. Inferencia Estadística La inferencia estadística puede dividirse en dos áreas principales: Estimación de Parámetros Prueba de Hipótesis Estimación de
Estimación de Parámetros. Jhon Jairo Padilla A., PhD.
 Estimación de Parámetros Jhon Jairo Padilla A., PhD. Inferencia Estadística La inferencia estadística puede dividirse en dos áreas principales: Estimación de Parámetros Prueba de Hipótesis Estimación de
Estimación de Parámetros Jhon Jairo Padilla A., PhD. Inferencia Estadística La inferencia estadística puede dividirse en dos áreas principales: Estimación de Parámetros Prueba de Hipótesis Estimación de
EVALUACION DEL EFECTO DE LA EVOLUCIÓN HETEROGENEA SITIO- ESPECIFICA SOBRE LA RECONSTRUCCIÓN FILOGENETICA MEDIANTE PARSIMONIA
 EVALUACION DEL EFECTO DE LA EVOLUCIÓN HETEROGENEA SITIO- ESPECIFICA SOBRE LA RECONSTRUCCIÓN FILOGENETICA MEDIANTE PARSIMONIA INTRODUCCION La biología comparada estudia la diversidad de especies analizando
EVALUACION DEL EFECTO DE LA EVOLUCIÓN HETEROGENEA SITIO- ESPECIFICA SOBRE LA RECONSTRUCCIÓN FILOGENETICA MEDIANTE PARSIMONIA INTRODUCCION La biología comparada estudia la diversidad de especies analizando
LAB 1 LAB 1/2. Estadística descriptiva. Pruebas de neutralidad. Mismatch distribution. Tajima Fu MacDonald-Kreitman
 LAB 1 Estadística descriptiva Pruebas de neutralidad Tajima Fu MacDonald-Kreitman Mismatch distribution LAB 1/2 1-numero de sitios segregantes en una muestra numero de sitios Tajima, 1989 : Var se puede
LAB 1 Estadística descriptiva Pruebas de neutralidad Tajima Fu MacDonald-Kreitman Mismatch distribution LAB 1/2 1-numero de sitios segregantes en una muestra numero de sitios Tajima, 1989 : Var se puede
Métodos de alineamiento. Bioinformática, Elvira Mayordomo
 Métodos de alineamiento Bioinformática, 24-2-16 Elvira Mayordomo Motivación: 2 razones para comparar secuencias biológicas 1. Los errores y omisiones en los datos biológicos producidos en la extracción
Métodos de alineamiento Bioinformática, 24-2-16 Elvira Mayordomo Motivación: 2 razones para comparar secuencias biológicas 1. Los errores y omisiones en los datos biológicos producidos en la extracción
Introducción a los árboles. Lección 11
 Introducción a los árboles Lección 11 Árbol: Conjunto de elementos de un mismo tipo, denominados nodos, que pueden representarse en un grafo no orientado, conexo y acíclico, en el que existe un vértice
Introducción a los árboles Lección 11 Árbol: Conjunto de elementos de un mismo tipo, denominados nodos, que pueden representarse en un grafo no orientado, conexo y acíclico, en el que existe un vértice
INSTITUTO NACIONAL DE ESTADÍSTICAS (INE) 29 de Abril de 2016
 29 de Abril de 2016") ANEXO ESTADÍSTICO 1 : COEFICIENTES DE VARIACIÓN Y ERROR ASOCIADO AL ESTIMADOR ENCUESTA NACIONAL DE EMPLEO (ENE) INSTITUTO NACIONAL DE ESTADÍSTICAS (INE) 9 de Abril de 016 1 Este anexo estadístico es una
ANEXO ESTADÍSTICO 1 : COEFICIENTES DE VARIACIÓN Y ERROR ASOCIADO AL ESTIMADOR ENCUESTA NACIONAL DE EMPLEO (ENE) INSTITUTO NACIONAL DE ESTADÍSTICAS (INE) 9 de Abril de 016 1 Este anexo estadístico es una
Introducción a Aprendizaje no Supervisado
 Introducción a Aprendizaje no Supervisado Felipe Suárez, Álvaro Riascos 25 de abril de 2017 2 / 33 Contenido 1. Motivación 2. k-medias Algoritmos Implementación 3. Definición 4. Motivación 5. Aproximación
Introducción a Aprendizaje no Supervisado Felipe Suárez, Álvaro Riascos 25 de abril de 2017 2 / 33 Contenido 1. Motivación 2. k-medias Algoritmos Implementación 3. Definición 4. Motivación 5. Aproximación
Métodos de alineamiento (3) Bioinformática, Elvira Mayordomo
 Bioinformática, Elvira Mayordomo") Métodos de alineamiento (3) Bioinformática, 7-3-16 Elvira Mayordomo Hoy veremos Multialineamiento (MSA): Un problema NP-completo Algoritmo aproximado para Multialineamiento: el método de la estrella Métodos
Métodos de alineamiento (3) Bioinformática, 7-3-16 Elvira Mayordomo Hoy veremos Multialineamiento (MSA): Un problema NP-completo Algoritmo aproximado para Multialineamiento: el método de la estrella Métodos
Congruencia- Árboles de consenso Soporte de grupos
 Congruencia- Árboles de consenso Soporte de grupos Los agrupamientos obtenidos a partir de análisis filogenéticos basados en distintos sets o conjuntos de datos son con frecuencia incongruentes. Comparaciones
Congruencia- Árboles de consenso Soporte de grupos Los agrupamientos obtenidos a partir de análisis filogenéticos basados en distintos sets o conjuntos de datos son con frecuencia incongruentes. Comparaciones
Estadística Demográfica. Grado en Relaciones Laborales y Recursos Humanos. Parte VI Tabla de mortalidad poblacional de momento: esperanza de vida
 Parte VI Tabla de mortalidad poblacional de momento: esperanza de vida 1 Vamos a suponer que el comportamiento poblacional en lo que se refiere al fenómeno mortalidad en la generación de t (análisis longitudinal),
Parte VI Tabla de mortalidad poblacional de momento: esperanza de vida 1 Vamos a suponer que el comportamiento poblacional en lo que se refiere al fenómeno mortalidad en la generación de t (análisis longitudinal),
Apellidos:... Nombre:...
 Apellidos:....................................... Nombre:........................................ Introducción a la Inteligencia Artificial 1 er Parcial de Teoría 12 Noviembre 2004 Ejercicio 1: Responder
Apellidos:....................................... Nombre:........................................ Introducción a la Inteligencia Artificial 1 er Parcial de Teoría 12 Noviembre 2004 Ejercicio 1: Responder
1. Método general. 2. Análisis de tiempos de ejecución. 3. Ejemplos de aplicación Problema de las 8 reinas Problema de la mochila 0/1.
 Backtracking. Método general. 2. Análisis de tiempos de ejecución. 3. Ejemplos de aplicación. 3.. Problema de las 8 reinas. 3.2. Problema de la mochila 0/. Método general El backtracking (método de retroceso
Backtracking. Método general. 2. Análisis de tiempos de ejecución. 3. Ejemplos de aplicación. 3.. Problema de las 8 reinas. 3.2. Problema de la mochila 0/. Método general El backtracking (método de retroceso
El TAD Árbol. El TAD Árbol
 Objetivos! Presentar el árbol como estructura de datos jerárquica! Estudiar diferentes variantes de árboles, tanto en su especificación como en su implementación Contenidos 3.1 Concepto, definiciones y
Objetivos! Presentar el árbol como estructura de datos jerárquica! Estudiar diferentes variantes de árboles, tanto en su especificación como en su implementación Contenidos 3.1 Concepto, definiciones y
Estructuras de Datos y Algoritmos
 Estructuras de Datos y Algoritmos Tema 5.1. Árboles. Árboles binarios y generales Prof. Dr. P. Javier Herrera Contenido 1. Introducción 2. Terminología 3. Árboles binarios 4. Árboles generales Tema 5.1.
Estructuras de Datos y Algoritmos Tema 5.1. Árboles. Árboles binarios y generales Prof. Dr. P. Javier Herrera Contenido 1. Introducción 2. Terminología 3. Árboles binarios 4. Árboles generales Tema 5.1.
INDICE Prefacio Como usar este libro Capitulo 1. Introducción Capitulo 2. Análisis exploratorio de los datos
 INDICE Prefacio Como usar este libro Capitulo 1. Introducción 1 El comienzo de todo: determinación lo que se debe saber 2 Evaluación numérica de las unidades de observación con la ayuda de las escalas
INDICE Prefacio Como usar este libro Capitulo 1. Introducción 1 El comienzo de todo: determinación lo que se debe saber 2 Evaluación numérica de las unidades de observación con la ayuda de las escalas
Análisis Filogenético. Reconstrucción de las relaciones genealógicas entre taxa
 Análisis Filogenético Reconstrucción de las relaciones genealógicas entre taxa Una de las diferencias entre las teorías evolutivas de Lamarck y Darwin esta relacionada al origen de las especies. Mientras
Análisis Filogenético Reconstrucción de las relaciones genealógicas entre taxa Una de las diferencias entre las teorías evolutivas de Lamarck y Darwin esta relacionada al origen de las especies. Mientras
ANÁLISIS DE DATOS. Jesús García Herrero
 ANÁLISIS DE DATOS Jesús García Herrero ANALISIS DE DATOS EJERCICIOS Una empresa de seguros de automóviles quiere utilizar los datos sobre sus clientes para obtener reglas útiles que permita clasificar
ANÁLISIS DE DATOS Jesús García Herrero ANALISIS DE DATOS EJERCICIOS Una empresa de seguros de automóviles quiere utilizar los datos sobre sus clientes para obtener reglas útiles que permita clasificar
Búsqueda Informada. Heurísticas
 Búsqueda Informada Heurísticas Búsqueda informada: heurística Ejemplo de heurística para el problema del viajante de comercio Clasificación de heurísticas Ventajas de las heurísticas Aplicando heurísticas
Búsqueda Informada Heurísticas Búsqueda informada: heurística Ejemplo de heurística para el problema del viajante de comercio Clasificación de heurísticas Ventajas de las heurísticas Aplicando heurísticas
OPTIMIZACIÓN Y SIMULACIÓN PARA LA EMPRESA. Tema 5 Simulación
 OPTIMIZACIÓN Y SIMULACIÓN PARA LA EMPRESA Tema 5 Simulación ORGANIZACIÓN DEL TEMA Sesiones: Introducción Ejemplos prácticos Procedimiento y evaluación de resultados INTRODUCCIÓN Simulación: Procedimiento
OPTIMIZACIÓN Y SIMULACIÓN PARA LA EMPRESA Tema 5 Simulación ORGANIZACIÓN DEL TEMA Sesiones: Introducción Ejemplos prácticos Procedimiento y evaluación de resultados INTRODUCCIÓN Simulación: Procedimiento
Estimar la adecuación de un nodo para ser expandido.
 Universidad Rey Juan Carlos Curso 2014 2015 Hoja de Problemas Tema 3 - Solución 1. Contesta a las siguientes preguntas: (a) Cuál es el objetivo de una función heurística aplicada a la búsqueda en el espacio
Universidad Rey Juan Carlos Curso 2014 2015 Hoja de Problemas Tema 3 - Solución 1. Contesta a las siguientes preguntas: (a) Cuál es el objetivo de una función heurística aplicada a la búsqueda en el espacio
Definición 1: Un grafo G es una terna ordenada (V(G), E(G), Ψ
, E(G), Ψ") Título: Un Arbol Natural Autor: Luis R. Morera onzález Resumen En este artículo se crea un modelo para representar los números naturales mediante un grafo, el cual consiste de de un árbol binario completo
Título: Un Arbol Natural Autor: Luis R. Morera onzález Resumen En este artículo se crea un modelo para representar los números naturales mediante un grafo, el cual consiste de de un árbol binario completo
IN34A - Optimización
 IN34A - Optimización Complejidad Leonardo López H. lelopez@ing.uchile.cl Primavera 2008 1 / 33 Contenidos Problemas y Procedimientos de solución Problemas de optimización v/s problemas de decisión Métodos,
IN34A - Optimización Complejidad Leonardo López H. lelopez@ing.uchile.cl Primavera 2008 1 / 33 Contenidos Problemas y Procedimientos de solución Problemas de optimización v/s problemas de decisión Métodos,
Biología Molecular y Filogenia en Micología
 Biología Molecular y Filogenia en Micología Biól. Nicolás Pastor Micología 2014 Por qué usar herramientas moleculares? Sistemática - Delimitación de especies Concepto filogenético de especie Diagnosis
Biología Molecular y Filogenia en Micología Biól. Nicolás Pastor Micología 2014 Por qué usar herramientas moleculares? Sistemática - Delimitación de especies Concepto filogenético de especie Diagnosis
Alineamiento de pares de secuencias
 Dr. Eduardo A. RODRÍGUEZ TELLO CINVESTAV-Tamaulipas 30 de mayo del 2013 Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Alineamiento de pares de secuencias 30 de mayo del 2013 1 / 61 1 Alineamiento de pares de secuencias
Dr. Eduardo A. RODRÍGUEZ TELLO CINVESTAV-Tamaulipas 30 de mayo del 2013 Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Alineamiento de pares de secuencias 30 de mayo del 2013 1 / 61 1 Alineamiento de pares de secuencias
Señales en el ADN. Bioinformática, Kevin Yip-CSE-CUHK (Universidad china de Hong-Kong)
") Señales en el ADN Bioinformática, 5-4-17 Kevin Yip-CSE-CUHK (Universidad china de Hong-Kong) Señales en el ADN Hemos visto cómo determinar y comparar secuencias de ADN Cómo podemos averiguar el significado
Señales en el ADN Bioinformática, 5-4-17 Kevin Yip-CSE-CUHK (Universidad china de Hong-Kong) Señales en el ADN Hemos visto cómo determinar y comparar secuencias de ADN Cómo podemos averiguar el significado
1. Diseñe algoritmos que permitan resolver eficientemente el problema de la mochila 0/1 para los siguientes casos:
 PROGRAMACIÓN DINÁMICA RELACIÓN DE EJERCICIOS Y PROBLEMAS 1. Diseñe algoritmos que permitan resolver eficientemente el problema de la mochila /1 para los siguientes casos: a. Mochila de capacidad W=15:
PROGRAMACIÓN DINÁMICA RELACIÓN DE EJERCICIOS Y PROBLEMAS 1. Diseñe algoritmos que permitan resolver eficientemente el problema de la mochila /1 para los siguientes casos: a. Mochila de capacidad W=15:
Secuenciación de DNA. Bioinformática, Elvira Mayordomo
 Secuenciación de DNA Bioinformática, 15-3-16 Elvira Mayordomo Hoy Introducción a la secuenciación de DNA El proyecto del genoma humano El proyecto de los 1000 genomas Mapa físico Sitios de restricción
Secuenciación de DNA Bioinformática, 15-3-16 Elvira Mayordomo Hoy Introducción a la secuenciación de DNA El proyecto del genoma humano El proyecto de los 1000 genomas Mapa físico Sitios de restricción
ÁRBOLES DE SINTAXIS. Los nodos no terminales (nodos interiores) están rotulados por los símbolos no terminales.
 están rotulados por los símbolos no terminales.") ÁRBOLES DE SINTAXIS ÁRBOL grafo dirigido acíclico. Los nodos no terminales (nodos interiores) están rotulados por los símbolos no terminales. Los nodos terminales (nodos hojas) están rotulados por los
ÁRBOLES DE SINTAXIS ÁRBOL grafo dirigido acíclico. Los nodos no terminales (nodos interiores) están rotulados por los símbolos no terminales. Los nodos terminales (nodos hojas) están rotulados por los
Ampliación de Algoritmos y Estructura de Datos Curso 02/03. Ejercicios
 272. En un problema determinado, una solución está dada por una tupla de n elementos (x, x 2,..., x n ). Para cada elemento existen en total m posibles valores. Comparar el número de nodos generados para
272. En un problema determinado, una solución está dada por una tupla de n elementos (x, x 2,..., x n ). Para cada elemento existen en total m posibles valores. Comparar el número de nodos generados para
Taxonomía y clasificación
 Taxonomía y clasificación Taxonomía vs. sistemática La Taxonomía es la ciencia que encargada de nombrar y clasificar los organismos. Sistemática es la ciencia que se encarga de determinar las relaciones
Taxonomía y clasificación Taxonomía vs. sistemática La Taxonomía es la ciencia que encargada de nombrar y clasificar los organismos. Sistemática es la ciencia que se encarga de determinar las relaciones
UNIDAD Creación del Árbol Octal de un objeto sólido vía la definición CSG
 UNIDAD 3 3 Creación de Árbol Octal vía la definición CSG 3.1 Creación del Árbol Octal de un objeto sólido vía la definición CSG Un árbol CSG hace uso de un conjunto de primitivas sólidas, estas primitivas
UNIDAD 3 3 Creación de Árbol Octal vía la definición CSG 3.1 Creación del Árbol Octal de un objeto sólido vía la definición CSG Un árbol CSG hace uso de un conjunto de primitivas sólidas, estas primitivas
Cap 7 Intervalos de Confianza
 Cap 7 Intervalos de Confianza Mate 3015 7.1-1 INTERVALOS DE CONFIANZA PARA UNA PROPORCIÓN 7.1-2 Estadísticas inferencial Ahora discutimos estadística inferencial -el proceso de generalizar la información
Cap 7 Intervalos de Confianza Mate 3015 7.1-1 INTERVALOS DE CONFIANZA PARA UNA PROPORCIÓN 7.1-2 Estadísticas inferencial Ahora discutimos estadística inferencial -el proceso de generalizar la información
Maestría en Bioinformática Probabilidad y Estadística: Clase 13
 Maestría en Bioinformática Probabilidad y Estadística: Clase 13 Gustavo Guerberoff gguerber@fing.edu.uy Facultad de Ingeniería Universidad de la República Mayo de 2010 Contenidos 1 Hidden Markov Models
Maestría en Bioinformática Probabilidad y Estadística: Clase 13 Gustavo Guerberoff gguerber@fing.edu.uy Facultad de Ingeniería Universidad de la República Mayo de 2010 Contenidos 1 Hidden Markov Models
259. El número de combinaciones de m objetos entre un conjunto de n, denotado por n, para n 1 y 0 m n, se puede definir recursivamente por: m
 258. Aplicar el algoritmo de programación dinámica para el problema del cambio de monedas sobre el siguiente ejemplo: n = 3, P = 9, c = (1, 3, 4). Qué ocurre si multiplicamos P y c por un valor constante,
258. Aplicar el algoritmo de programación dinámica para el problema del cambio de monedas sobre el siguiente ejemplo: n = 3, P = 9, c = (1, 3, 4). Qué ocurre si multiplicamos P y c por un valor constante,
Demostrando cotas inferiores: Arboles de decisión
 Demostrando cotas inferiores: Arboles de decisión De la misma forma que la técnica basada en la mejor estrategia del adversario, vamos a utilizar los árboles de decisión para establecer una cota inferior
Demostrando cotas inferiores: Arboles de decisión De la misma forma que la técnica basada en la mejor estrategia del adversario, vamos a utilizar los árboles de decisión para establecer una cota inferior
Pablo Vinuesa 2007, 1
 Curso fundamenteal de Inferencia Filogenética Molecular Pablo Vinuesa (vinuesa@ccg.unam.mx) Progama de Ingeniería Genómica, CCG, UNAM http://www.ccg.unam.mx/~vinuesa/ Tutor: PDCBM, Ciencias Biológicas,
Curso fundamenteal de Inferencia Filogenética Molecular Pablo Vinuesa (vinuesa@ccg.unam.mx) Progama de Ingeniería Genómica, CCG, UNAM http://www.ccg.unam.mx/~vinuesa/ Tutor: PDCBM, Ciencias Biológicas,
ECONOMETRÍA II Prof.: Begoña Álvarez TEMA 1 INTRODUCCIÓN. Estimación por máxima verosimilitud y conceptos de teoría asintótica
 ECONOMETRÍA II Prof.: Begoña Álvarez 2007-2008 TEMA 1 INTRODUCCIÓN Estimación por máxima verosimilitud y conceptos de teoría asintótica 1. ESTIMACIÓN POR MÁXIMA VEROSIMILITUD (MAXIMUM LIKELIHOOD) La estimación
ECONOMETRÍA II Prof.: Begoña Álvarez 2007-2008 TEMA 1 INTRODUCCIÓN Estimación por máxima verosimilitud y conceptos de teoría asintótica 1. ESTIMACIÓN POR MÁXIMA VEROSIMILITUD (MAXIMUM LIKELIHOOD) La estimación
JUNIO Bloque A
 Selectividad Junio 009 JUNIO 009 Bloque A 1.- Estudia el siguiente sistema en función del parámetro a. Resuélvelo siempre que sea posible, dejando las soluciones en función de parámetros si fuera necesario.
Selectividad Junio 009 JUNIO 009 Bloque A 1.- Estudia el siguiente sistema en función del parámetro a. Resuélvelo siempre que sea posible, dejando las soluciones en función de parámetros si fuera necesario.
Tipos algebraicos y abstractos. Algoritmos y Estructuras de Datos I. Tipos algebraicos
 Algoritmos y Estructuras de Datos I 1 cuatrimestre de 009 Departamento de Computación - FCEyN - UBA Programación funcional - clase Tipos algebraicos Tipos algebraicos y abstractos ya vimos los tipos básicos
Algoritmos y Estructuras de Datos I 1 cuatrimestre de 009 Departamento de Computación - FCEyN - UBA Programación funcional - clase Tipos algebraicos Tipos algebraicos y abstractos ya vimos los tipos básicos
Una pregunta pendiente
 Una pregunta pendiente Cómo podemos construir un generador (casi) uniforme para una relación? Recuerde el problema KS definido en la sección anterior y la relación: R KS = {((~a, b), ~x) ~a 2 N n y ~x
Una pregunta pendiente Cómo podemos construir un generador (casi) uniforme para una relación? Recuerde el problema KS definido en la sección anterior y la relación: R KS = {((~a, b), ~x) ~a 2 N n y ~x
Árboles y esquemas algorítmicos. Tema III
 Árboles y esquemas algorítmicos Tema III Bibliografía Tema III (lecciones 15 a 22) del libro Campos Laclaustra, J.: Estructuras de Datos y Algoritmos, Prensas Universitarias de Zaragoza, Colección Textos
Árboles y esquemas algorítmicos Tema III Bibliografía Tema III (lecciones 15 a 22) del libro Campos Laclaustra, J.: Estructuras de Datos y Algoritmos, Prensas Universitarias de Zaragoza, Colección Textos
Econometría II. Tema 1: Revisión del Modelo de Regresión Múltiple Ejercicios
 Econometría II Tema 1: Revisión del Modelo de Regresión Múltiple Ejercicios 1. Problema En el chero "Produccio.xls" se presenta la información sobre la producción Y, trabajo X 2 y capital X 3 en el sector
Econometría II Tema 1: Revisión del Modelo de Regresión Múltiple Ejercicios 1. Problema En el chero "Produccio.xls" se presenta la información sobre la producción Y, trabajo X 2 y capital X 3 en el sector
Ejercicios Propuestos de Métodos de Búsqueda
 Inteligencia rtificial Ejercicios de Métodos de úsqueda Ejercicios Propuestos de Métodos de úsqueda os siguientes ejercicios, corresponden a la práctica de Métodos de úsqueda y son propuestos a los alumnos
Inteligencia rtificial Ejercicios de Métodos de úsqueda Ejercicios Propuestos de Métodos de úsqueda os siguientes ejercicios, corresponden a la práctica de Métodos de úsqueda y son propuestos a los alumnos
1/26/11! Taller Latinoamericano de Evolución Molecular! 2011! Relojes Moleculares:! Fechación de Clados con Datos Moleculares! EL RELOJ MOLECULAR!
 Taller Latinoamericano de Evolución Molecular! 2011! Relojes Moleculares:! Fechación de Clados con Datos Moleculares! Prof. Susana Magallón! Enero 27, 2011! EL RELOJ MOLECULAR! La distancia genética entre
Taller Latinoamericano de Evolución Molecular! 2011! Relojes Moleculares:! Fechación de Clados con Datos Moleculares! Prof. Susana Magallón! Enero 27, 2011! EL RELOJ MOLECULAR! La distancia genética entre
Capítulo 8. Árboles. Continuar
 Capítulo 8. Árboles Continuar Introducción Uno de los problemas principales para el tratamiento de los grafos es que no guardan una estructura establecida y que no respetan reglas, ya que la relación entre
Capítulo 8. Árboles Continuar Introducción Uno de los problemas principales para el tratamiento de los grafos es que no guardan una estructura establecida y que no respetan reglas, ya que la relación entre
Búsqueda en línea y Búsqueda multiagente
 Búsqueda en línea y Búsqueda multiagente Ingeniería Informática, 4º Curso académico: 2011/2012 Profesores: Ramón Hermoso y Matteo Vasirani 1 Tema 2: Agentes basados en Búsqueda Resumen: 2. Agentes basados
Búsqueda en línea y Búsqueda multiagente Ingeniería Informática, 4º Curso académico: 2011/2012 Profesores: Ramón Hermoso y Matteo Vasirani 1 Tema 2: Agentes basados en Búsqueda Resumen: 2. Agentes basados
Tema 6: Introducción a la Inferencia Bayesiana
 Tema 6: Introducción a la Inferencia Bayesiana Conchi Ausín Departamento de Estadística Universidad Carlos III de Madrid concepcion.ausin@uc3m.es CESGA, Noviembre 2012 Contenidos 1. Elementos básicos de
Tema 6: Introducción a la Inferencia Bayesiana Conchi Ausín Departamento de Estadística Universidad Carlos III de Madrid concepcion.ausin@uc3m.es CESGA, Noviembre 2012 Contenidos 1. Elementos básicos de
Estimación. Introducción. Sea X la variable aleatoria poblacional con distribución de probabilidad f θ donde. es el parámetro poblacional desconocido
 Tema : Introducción a la Teoría de la Estimación Introducción Sea X la variable aleatoria poblacional con distribución de probabilidad f θ (x), donde θ Θ es el parámetro poblacional desconocido Objetivo:
Tema : Introducción a la Teoría de la Estimación Introducción Sea X la variable aleatoria poblacional con distribución de probabilidad f θ (x), donde θ Θ es el parámetro poblacional desconocido Objetivo:
Muchas de las ecuaciones de recurrencia que vamos a usar en este curso tienen la siguiente forma: ( c n =0 T (n) = a T (b n b.
 = a T (b n b.") El Teorema Maestro Muchas de las ecuaciones de recurrencia que vamos a usar en este curso tienen la siguiente forma: ( c n =0 T (n) = a T (b n b c)+f (n) n 1 donde a, b y c son constantes, y f (n) es una
El Teorema Maestro Muchas de las ecuaciones de recurrencia que vamos a usar en este curso tienen la siguiente forma: ( c n =0 T (n) = a T (b n b c)+f (n) n 1 donde a, b y c son constantes, y f (n) es una
Slide 1 / 68. Slide 2 / 68. Slide 3 / Qué es una especie? 2 Qué debe suceder en una población para que ocurra especiación?
 1 Qué es una especie? Slide 1 / 68 2 Qué debe suceder en una población para que ocurra especiación? Slide 2 / 68 3 Enumera y describe cuatro tipos de aislamiento reproductivo. Slide 3 / 68 4 Qué es el
1 Qué es una especie? Slide 1 / 68 2 Qué debe suceder en una población para que ocurra especiación? Slide 2 / 68 3 Enumera y describe cuatro tipos de aislamiento reproductivo. Slide 3 / 68 4 Qué es el
Tema 4: Alineamiento Múltiple y Filogenias (3)
") Bioinformática lásica Bioinformática lásica ema 4: lineamiento Múltiple y Filogenias (3) Sección 3: Filogenias Dr. Oswaldo relles Universidad de Málaga La filogenia estudia las relaciones de parentesco
Bioinformática lásica Bioinformática lásica ema 4: lineamiento Múltiple y Filogenias (3) Sección 3: Filogenias Dr. Oswaldo relles Universidad de Málaga La filogenia estudia las relaciones de parentesco
Capítulo 5. Conclusiones
 Capítulo 5 Conclusiones En este trabajo se desarrolló un sistema capaz de clasificar enunciados dependiendo de la opinión que cada uno expresa acerca de una película. Se cumplió entonces con el objetivo
Capítulo 5 Conclusiones En este trabajo se desarrolló un sistema capaz de clasificar enunciados dependiendo de la opinión que cada uno expresa acerca de una película. Se cumplió entonces con el objetivo
ASIGNATURA: (TIS-106) Estructuras de Datos II DOCENTE: Ing. Freddy Melgar Algarañaz
 Estructuras de Datos II DOCENTE: Ing. Freddy Melgar Algarañaz") TEMA 1. Árboles Generalizados Son estructuras de datos no lineales, o también denominadas estructuras multienlazadas. El árbol es una estructura de datos fundamental en informática, muy utilizada en todos
TEMA 1. Árboles Generalizados Son estructuras de datos no lineales, o también denominadas estructuras multienlazadas. El árbol es una estructura de datos fundamental en informática, muy utilizada en todos
Códigos IRA. Máster en Multimedia y Comunicaciones Comunicaciones Digitales. Luca Martino
 Códigos IRA Máster en Multimedia y Comunicaciones Comunicaciones Digitales Luca Martino Codificación de Canal! Supongamos tener un canal binario discreto, simétrico sin memoria:! Objetivo: encontrar una
Códigos IRA Máster en Multimedia y Comunicaciones Comunicaciones Digitales Luca Martino Codificación de Canal! Supongamos tener un canal binario discreto, simétrico sin memoria:! Objetivo: encontrar una
Aprendizaje Computacional y Extracción de Información
 Aprendizaje Computacional y Extracción de Información Inferencia Gramatical Jose Oncina oncina@dlsi.ua.es Dep. Lenguajes y Sistemas Informáticos Universidad de Alicante 26 de septiembre de 2007 J. Oncina
Aprendizaje Computacional y Extracción de Información Inferencia Gramatical Jose Oncina oncina@dlsi.ua.es Dep. Lenguajes y Sistemas Informáticos Universidad de Alicante 26 de septiembre de 2007 J. Oncina
Formulación del problema de la ruta más corta en programación lineal
 Formulación del problema de la ruta más corta en programación lineal En esta sección se describen dos formulaciones de programación lineal para el problema de la ruta más corta. Las formulaciones son generales,
Formulación del problema de la ruta más corta en programación lineal En esta sección se describen dos formulaciones de programación lineal para el problema de la ruta más corta. Las formulaciones son generales,
TAXONOMÍA MOLECULAR TAXONOMÍA MOLECULAR
 TAXONOMÍA MOLECULAR TAXONOMÍA MOLECULAR Microbiología a General Área Micología Dra. Alicia Luque CEREMIC Taxonomía clásica Carecen de reproducibilidad: los caracteres fenotípicos pueden variar con las
TAXONOMÍA MOLECULAR TAXONOMÍA MOLECULAR Microbiología a General Área Micología Dra. Alicia Luque CEREMIC Taxonomía clásica Carecen de reproducibilidad: los caracteres fenotípicos pueden variar con las
A B MIN C D E F MAX x E.T.S.I. INFORMÁTICA 4º CURSO. INTELIGENCIA ARTIFICIAL E INGENIERÍA DEL CONOCIMIENTO
 E.T.S.I. INFORMÁTICA 4º CURSO. INTELIGENCIA ARTIFICIAL E INGENIERÍA DEL CONOCIMIENTO UNIVERSIDAD DE MÁLAGA Dpto. Lenguajes y Ciencias de la Computación RELACIÓN DE PROBLEMAS. TEMA IV. PROBLEMAS DE JUEGOS.
E.T.S.I. INFORMÁTICA 4º CURSO. INTELIGENCIA ARTIFICIAL E INGENIERÍA DEL CONOCIMIENTO UNIVERSIDAD DE MÁLAGA Dpto. Lenguajes y Ciencias de la Computación RELACIÓN DE PROBLEMAS. TEMA IV. PROBLEMAS DE JUEGOS.
Ideas básicas del diseño experimental
 Ideas básicas del diseño experimental Capítulo 4 de Analysis of Messy Data. Milliken y Johnson (1992) Diseño de experimentos p. 1/23 Ideas básicas del diseño experimental Antes de llevar a cabo un experimento,
Ideas básicas del diseño experimental Capítulo 4 de Analysis of Messy Data. Milliken y Johnson (1992) Diseño de experimentos p. 1/23 Ideas básicas del diseño experimental Antes de llevar a cabo un experimento,
1 Introducción. 2 Modelo. Hipótesis del modelo MODELO DE REGRESIÓN LOGÍSTICA
 MODELO DE REGRESIÓN LOGÍSTICA Introducción A grandes rasgos, el objetivo de la regresión logística se puede describir de la siguiente forma: Supongamos que los individuos de una población pueden clasificarse
MODELO DE REGRESIÓN LOGÍSTICA Introducción A grandes rasgos, el objetivo de la regresión logística se puede describir de la siguiente forma: Supongamos que los individuos de una población pueden clasificarse
No se permiten libros ni apuntes. Ejercicio 1 Ejercicio 2 Ejercicio 3 Ejercicio 4 TOTAL NOTA
 Junio Duración: h Ejercicio Ejercicio Ejercicio Ejercicio TOTAL NOTA Ejercicio : [ puntos: respuesta acertada = +., respuesta incorrecta =.] Complete las siguientes frases y conteste a cada una con verdadero
Junio Duración: h Ejercicio Ejercicio Ejercicio Ejercicio TOTAL NOTA Ejercicio : [ puntos: respuesta acertada = +., respuesta incorrecta =.] Complete las siguientes frases y conteste a cada una con verdadero
Matemáticas Básicas para Computación
 Matemáticas Básicas para Computación MATEMÁTICAS BÁSICAS PARA COMPUTACIÓN 1 Sesión No. 11 Nombre: Árboles Objetivo: Al término de la sesión el participante conocerá los tipos de grafos específicamente
Matemáticas Básicas para Computación MATEMÁTICAS BÁSICAS PARA COMPUTACIÓN 1 Sesión No. 11 Nombre: Árboles Objetivo: Al término de la sesión el participante conocerá los tipos de grafos específicamente
ESTIMACIÓN Y PRUEBA DE HIPÓTESIS INTERVALOS DE CONFIANZA
 www.jmontenegro.wordpress.com UNI ESTIMACIÓN Y PRUEBA DE HIPÓTESIS INTERVALOS DE CONFIANZA PROF. JOHNNY MONTENEGRO MOLINA Objetivos Desarrollar el concepto de estimación de parámetros Explicar qué es una
www.jmontenegro.wordpress.com UNI ESTIMACIÓN Y PRUEBA DE HIPÓTESIS INTERVALOS DE CONFIANZA PROF. JOHNNY MONTENEGRO MOLINA Objetivos Desarrollar el concepto de estimación de parámetros Explicar qué es una
III Verano de Probabilidad y Estadística Curso de Procesos de Poisson (Víctor Pérez Abreu) Lista de Ejercicios
 Lista de Ejercicios") III Verano de Probabilidad y Estadística Curso de Procesos de Poisson (Víctor Pérez Abreu) Lista de Ejercicios Esta lista contiene ejercicios y problemas tanto teóricos como de modelación. El objetivo
III Verano de Probabilidad y Estadística Curso de Procesos de Poisson (Víctor Pérez Abreu) Lista de Ejercicios Esta lista contiene ejercicios y problemas tanto teóricos como de modelación. El objetivo
PRACTICA XI: ALINEAMIENTO POR TRADUCCION INVERSA Y ANALISIS FILOGENETICO POR EL CRITERIO DE DISTANCIAS
 PRACTICA XI: ALINEAMIENTO POR TRADUCCION INVERSA Y ANALISIS FILOGENETICO POR EL CRITERIO DE DISTANCIAS Objetivo: - Aplicar las técnicas para realizar el alineamiento de secuencias de DNA codificantes empleadas
PRACTICA XI: ALINEAMIENTO POR TRADUCCION INVERSA Y ANALISIS FILOGENETICO POR EL CRITERIO DE DISTANCIAS Objetivo: - Aplicar las técnicas para realizar el alineamiento de secuencias de DNA codificantes empleadas
Andrés M. Pinzón Centro de Bioinformática Instituto de Biotecnología Universidad Nacional de Colombia
 Alineamiento: Análisis computacional de secuencias Andrés M. Pinzón Centro de Bioinformática Instituto de Biotecnología Universidad Nacional de Colombia Por qué y para qué... Tengo una secuencia de DNA/Proteína......
Alineamiento: Análisis computacional de secuencias Andrés M. Pinzón Centro de Bioinformática Instituto de Biotecnología Universidad Nacional de Colombia Por qué y para qué... Tengo una secuencia de DNA/Proteína......
Tema 8: Árboles de Clasificación
 Tema 8: Árboles de Clasificación p. 1/11 Tema 8: Árboles de Clasificación Abdelmalik Moujahid, Iñaki Inza, Pedro Larrañaga Departamento de Ciencias de la Computación e Inteligencia Artificial Universidad
Tema 8: Árboles de Clasificación p. 1/11 Tema 8: Árboles de Clasificación Abdelmalik Moujahid, Iñaki Inza, Pedro Larrañaga Departamento de Ciencias de la Computación e Inteligencia Artificial Universidad
SIMULACION. Urna 1. s(x) h(x) Urna 2. s(x) Dado. Urna /6 2 1/6 3 1/6 4 1/6 5 1/6 6 1/6 Dado 1 1/6 2 1/6 3 1/6 4 1/6 5 1/6 6 1/6
 h(x) Urna 2. s(x) Dado. Urna /6 2 1/6 3 1/6 4 1/6 5 1/6 6 1/6 Dado 1 1/6 2 1/6 3 1/6 4 1/6 5 1/6 6 1/6") SIMULACION x p(x) x h(x) 6 4 5 2 3 Urna /6 2 /6 3 /6 4 /6 5 /6 6 /6 Dado /6 2 /6 3 /6 4 /6 5 /6 6 /6 x p(x) x h(x) s(x) 4 5 2 3 Urna 2 0.2 2 0.2 3 0.2 4 0.2 5 0.2 6 0.0 Dado /6.2 2 /6.2 3 /6.2 4 /6.2 5
SIMULACION x p(x) x h(x) 6 4 5 2 3 Urna /6 2 /6 3 /6 4 /6 5 /6 6 /6 Dado /6 2 /6 3 /6 4 /6 5 /6 6 /6 x p(x) x h(x) s(x) 4 5 2 3 Urna 2 0.2 2 0.2 3 0.2 4 0.2 5 0.2 6 0.0 Dado /6.2 2 /6.2 3 /6.2 4 /6.2 5
Procesos estocásticos Cadenas de Márkov
 Procesos estocásticos Cadenas de Márkov Curso: Investigación de Operaciones Ing. Javier Villatoro PROCESOS ESTOCASTICOS Procesos estocásticos Es un proceso o sucesión de eventos que se desarrolla en el
Procesos estocásticos Cadenas de Márkov Curso: Investigación de Operaciones Ing. Javier Villatoro PROCESOS ESTOCASTICOS Procesos estocásticos Es un proceso o sucesión de eventos que se desarrolla en el
2008 Pablo Vinuesa, 1
 Introducción a la Inferencia Filogenética y Evolución Molecular 23-26 Junio 2008, Fac. C. Biológicas - UANL Pablo Vinuesa (vinuesa@ccg.unam.mx) Centro de Ciencias Genómicas-UNAM, México http://www.ccg.unam.mx/~vinuesa/
Introducción a la Inferencia Filogenética y Evolución Molecular 23-26 Junio 2008, Fac. C. Biológicas - UANL Pablo Vinuesa (vinuesa@ccg.unam.mx) Centro de Ciencias Genómicas-UNAM, México http://www.ccg.unam.mx/~vinuesa/