Grupo de Estudio de Enfermedades Neuromusculares Sociedad Española de Neurología
|
|
|
- María Carmen Alarcón Silva
- hace 7 años
- Vistas:
Transcripción
1 1 NEUROPATIAS HEREDITARIAS Grupo de Estudio de Enfermedades Neuromusculares Sociedad Española de Neurología documento PDF creado para la web el 7 de Julio del 2004

2 2 Neuropatías hereditarias 1. Introducción Las neuropatías hereditarias incluyen una amplia serie de síndromes que aparecen recopilados en la tabla 1. Existen dos grandes categorías clínicas: la primera caracterizada por una semiología exclusiva o predominantemente polineurítica, mientras que en la segunda a esta semiología polineurítica se agregan manifestaciones por afectación de otros sistemas además del nervioso periférico (SNP). Indudablemente las formas polineuropáticas puras son las más frecuentes y dentro de éstas la mayor prevalencia corresponde a la enfermedad de Charcot-Marie-Tooth (CMT) también conocida con el epónimo de neuropatía sensitivo-motora hereditaria (NSMH), a la que prestaremos una especial atención en este manuscrito. 2. CMT Clásicamente CMT incluía formas de atrofia muscular peroneal con herencia autosómica dominante (AD), autosómica recesiva [(AR); conocida esta forma con el epónimo de enfermedad de Dejerine-Sottas (EDS)], o con herencia ligada al sexo. Se admitía también el caso esporádico si tras un meticuloso escrutinio diagnóstico se habían excluido causas adquiridas de polineuropatía.
 también conocida con el epónimo")
3 Los estudios de conducción nerviosa son esenciales para el diagnóstico de CMT. Se distinguen dos patrones neurofisiológicos fundamentales. El primero se caracteriza por una acusada, difusa y simétrica lentificación de las velocidades de conducción nerviosa (VCN) (<38 m/s para el nervio mediano). Precisamente esta afectación uniforme de los parámetros de conducción nerviosa distingue a CMT de otras polineuropatías adquiridas, en las que es característica la variabilidad en tiempo y espacio de las alteraciones neurofisiológicas. El segundo patrón neurofisiológico es el esperable en una neuropatía axonal, es decir, VCNs normales o ligeramente enlentecidas con potenciales distales de amplitud reducida. Cuando la pérdida de axones distales es muy acusada y hay una marcada caída de la amplitud de los potenciales distales, en estas formas axonales puede haber un descenso de la VCN hasta el rango desmielinizante. En tal circunstancia, sin embargo, el estudio de conducción proximal (p.ej., latencia de punto de Erb a músculo deltoides) demostrará las alteraciones propias de una neuropatía axonal. En CMT ligada al cromosoma X las alteraciones neurofisiológicas no siguen este esquema, de modo que puede observarse ya una neuropatía preferentemente axonal o bien de predominio desmielinizante, incluso en miembros de una misma familia. 3 La biopsia de nervio ha perdido terreno como técnica diagnóstico del enfermo con sospecha de CMT. De hecho, sólo está indicada en casos clínicos complejos, o con fines de investigación clínica y con el debido consentimiento

4 informado. En correspondencia con lo descrito en el párrafo anterior, el estudio histológico suele demostrar una neuropatía hipertrófica en las formas desmielinizantes, y una pérdida de fibras mielínicas gruesas con variable regeneración en las formas axonales. 4 En las dos últimas décadas se han identificado 18 loci localizados en 11 cromosomas diferentes. El defecto génico se ha identificado para tres proteínas de la mielina (PMP22 en el cromosoma 17; P0 en el cromosoma 1; y conexina 32 en el cromosoma X) y para un factor de transcripción (EGR2 en el cromosoma 10). Los defectos moleculares son ya una trisomía alélica de 1 5 Mb en el caso de CMT1A (cromosoma 17) o bien mutaciones puntiformes en cualquiera de los cuatro genes clonados. En la neuropatía por vulnerabilidad excesiva a la presión (NVEP) hay una delección de la región duplicada en CMT-1A. Dado que aquí el cuadro clinico de NVEP es específico (parálisis recurrente de los nervios presionados) y que rarísima vez cursa con un fenotipo de atrofia muscular peroneal, no haremos más comentario sobre esta entidad. Los estudios de genética molecular han permitido comprender la enorme heterogeneidad clínica CMT, es decir, el mismo fenotipo ocurre con mutaciones diferentes, y a su vez una misma mutación origina fenotipos de diversa severidad. Se ha demostrado que las mutaciones subyacentes pueden aparecer de novo, lo cual corrobora la realidad clínica de los clásicos CMT esporádicos. Aunque
 y para un factor de transcripción (EGR2 en el")
5 excepcionalmente, se ha comprobado que la expresión clínica de algunas mutaciones ocurren en situaciones de heterocigosidad (herencia AD) o sólo de homocigosidad (herencia AR). Y finalmente se ha demostrado cómo algunas mutaciones de proteínas de la mielina se traducen no en una mielinopatía sino en una axonopatía; este fenómeno es habitual para las mutaciones de conexina 32 y excepcional para las de P CMT: algoritmo diagnóstico Pese a todos los avances genéticos, en la práctica clínica el diagnóstico y clasificación de CMT debe partir de los datos clínicos, genealógicos y neurofisiológicos; sólo así es como los estudios de genética molecular permitirán una división etiopatogénica menos compleja que la expuesta en la tabla 1 (ver final del documento). Escapa al objeto de este texto revisar la clínica de CMT; bastará recordar que va desde formas subclínicas a graves síndromes polineuropáticos deficitarios con pie cavo. La ausencia de semiología sensitiva irritativa en CMT es un rasgo diferencial con las polineuropatías crónicas adquiridas donde tales síntomas ocurren asiduamente. Las formas más frecuentes de CMT son las que tienen transmisión AD (en torno al 80%), con una proporción comparable entre el tipo I (usualmente tipo 1A) y tipo II (ver tabla 1 al final del documento). Las formas ligadas al sexo representan un 15%; el 5% restante corresponde a otras formas de CMT recogidas en la tabla 1.

6 Nuestro esquema de trabajo se resume en el algoritmo de la figura 1(ver final del documento). No proceder así, esto es, la pretensión de llegar a un diagnóstico partiendo de los hallazgos genéticos de laboratorio, si bien excepcionalmente útil, es fuente de contínuos errores Neuropatías sensitivas o preferentemente sensitivas Cabe considerar aquí tanto las polineuropatías amiloidóticas familiares (PAF) como las neuropatías sensitivas hereditarias (NSH). (ver algoritmo en formato PDF en esta misma sección de la web) Las PAF se clasifican en función de las proteinas amiloidogénicas y de los defectos genéticos subyacentes (ver tabla 1). La transmisión es AD. Durante buena parte del curso clínico, la semiología predominante es sensitiva e incluye parestesias, disestesias, e hipoestesia de predominio termoalgésico (pseudosiringomielia). Es característico de las PAF el desarrollo tempranero de disautonomía (p.ej., diarrea, hipotensión ortostática, anhidrosis o impotencia). La paresia y amiotrofia de las piernas aparece más tardíamente. De forma variable puede agregarse otra semiología que incluye neuropatía craneal, miocardiopatía, opacidades del vítreo, síndrome del túnel carpiano o distrofia corneal. El estudio neurofisiológico pone de manifiesto los hallazgos propios de una neuropatía axonal. Cuando no hay información genealógica, la biopsia de nervio es esencial para demostrar los depósitos amiloides en el endoneuro y alrededor de los
 Las PAF se clasifican en función de las proteinas amiloidogénicas y de los defectos genéticos subyacentes (ver tabla 1).")
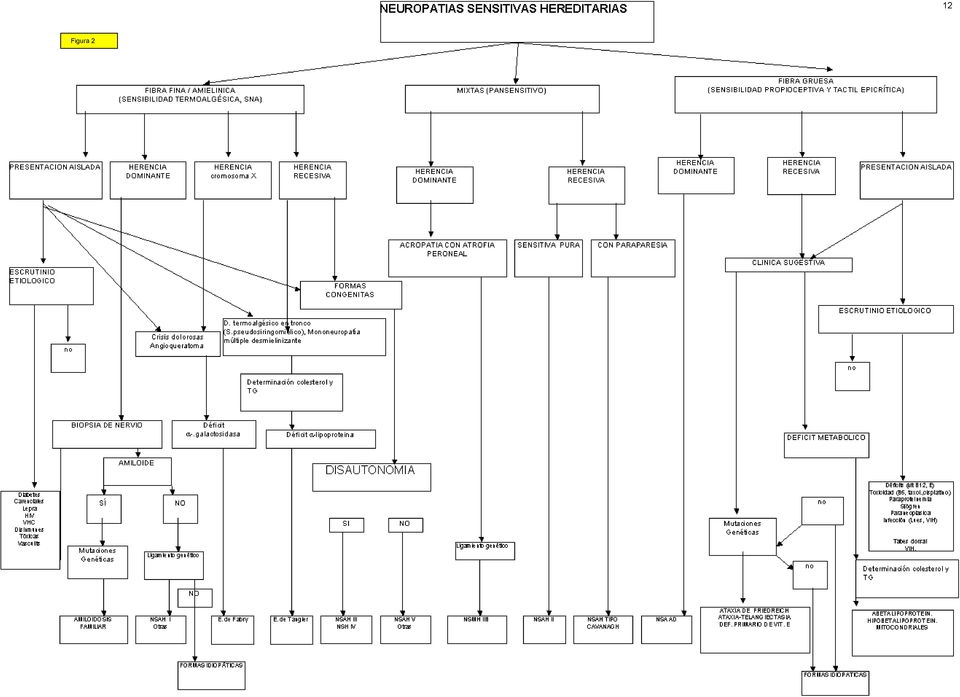
7 vasos sanguíneos (circunstancialmente esta alteración patológica puede detectarse en otros tejidos obviando la necesidad de la biopsia de nervio). Si se dispone de información genética familiar, puede irse directamente al diagnóstico molecular, que consiste en la detección de mutaciones en los genes de las proteínas amiloidogénicas (ver tabla 1). En zonas de riesgo de PAF, la epidemiología genética es de gran ayuda para orientar el estudio genético molecular, incluso en casos aparentemente esporádicos. 7 Las NSH incluyen cinco síndromes diferentes que aparecen recogidos en la tabla 1. Con transmisión AD o AR, el cuadro clínico se mueve entre una acropatía ulceromutilante con o sin componente tabético y una disautonomía congénita. Las alteraciones neurofisiológicas están circunscritas al sistema sensitivo, un hecho diferencial de las PAF. La biopsia de nervio demuestra una combinación variable de pérdida de fibras mielínicas gruesas y finas y amielínicas, que es característica para cada síndrome de NSH PAF y NSH: algoritmo diagnóstico El algoritmo diagnóstico (Figura 2 al final del documento) en este terreno se centra en la estrategia a seguir ante un paciente con un síndrome polineuropático sensitivo de larga evolución. Las dos posibilidades básicas es que se trate de un caso esporádico o con historia familiar positiva. En la primera situación deben excluirse causas

8 adquiridas de neuropatía atáxica o disautonómica. Una vez excluidas éstas y si la biopsia de nervio demuestra depósitos amiloides, se procede como en una PAF. Cuando la historia familiar es positiva, la forma de transmisión hereditaria, la semiología predominante y los hallazgos de los estudios neurofisiológicos y de la biopsia de nervio permiten la tipificación nosológica. Dada la rareza de las NSH no hemos entrado en el detalle de los hallazgos histológicos del nervio. Ni tampoco hemos incluido en el algoritmo los estudios de ligamiento genético que han permitido en familias aisladas la localización de un par de loci (ver tabla 1) Neuropatías hereditarias con semiología plus Como hemos esbozado antes, se incluyen aquí síndromes en los que la semiología polineuropática va acompañada de síntomas y signos de otros sistemas además del SNP (ver tabla 1). Para elaborar el algoritmo diagnóstico (figura 3) hemos partido de nuevo de los datos clínicogenéticos. La mayor parte de estos síndromes reconocen una transmisión AR y se encuadran entre las enfermedades por acúmulo lisosómico o entre las debidas a un trastorno de la reparación del ADN; en este grupo se encuadran también la ataxia de Friedreich y la neuropatía axonal gigante. En casos de herencia ligada al sexo el diagnóstico queda prácticamente circunscrito a la enfermedad de Fabry. En casos con herencia AD y neuropatía axonal las dos posibilidades básicas son ciertos tipos de ataxia dominante y las porfirias. Aunque no incluido en el algoritmo, debe
. 8 4.")
9 mencionarse la citopatía micocondrial (ver algoritmo al respecto de la miopatía mitocondrial). Evoca este trastorno un componente plus que incluya datos semiológicos de los síndromes mitocondriales (p.ej., MELAS, MERRF o LHON). Si este componente plus es claro puede irse directamente al estudio molecular en una muestra de sangre. Cuando éste es negativo o no hay un componente plus evocador, debe efectuarse biopsia muscular para el correspondiente estudio morfológico, bioquímico y molecular. 9 Referencias Las referencias citadas a continuación corresponden a trabajos de revisión de reciente aparición. 1. Lupsky JR. Recessive Charcot-Marie-Tooth disease. Ann Neurol 2000; 47: Nelis E, Haites N, Van Broeckhoven C. Mutations in the peripheral myelin genes and associated genes in the peripheral neuropathies. Hum Mut 1999; 13: Pareyson D. Charcot-Marie-Tooth disease and related neuropathies: molecular basis for distinction and diagnosis. Muscle Nerve 1999; 22: Reilly MR. Genetically determined neuropathies. J Neurol 1998; 245: 6-13.

10 Tabla 1. Clasificación de las neuropatías hereditarias* 1. Neuropatías cuya semiología polineurítica es la primaria o predominante 1.1. Neuropatía sensitivomotora hereditaria (NSMH) o enfermedad de Charcot-Marie-Tooth (CMT) sexo) - NSMH I (CMT 1) (transmisión usual AD o ligada al - NSMH IA (CMT 1A) Duplicación PMP-22 o mutaciones gen PMP22 (17p.11) - NSMH IB (CMT 1B) Mutaciones gen P0 (1q22) - NSMH IC (CMT 1C) Mutaciones gen EGR2 (10q) desconocida - NSMH ID (CMT 1D) Localización genética - CMT X1 Mutaciones gen conexina 32 (Xq13) - CMT X2 Localización exacta no establecida - Formas recesivas (ver CMT 4) NSMH II (CMT 2) (transmisión usual AD) - NSMH IIA (CMT 2A) 1p35-p36 - NSMH IIB (CMT 2B) 3q13-q22
 o enfermedad de Charcot-Marie-Tooth (CMT) sexo) - NSMH I (CMT 1) (transmisión usual AD o ligada al - NSMH IA (CMT 1A)")
11 desconocida - NSMH IIC (CMT 2C; Localización genética con parálisis cuerdas vocales) - NSMH IID (DMT 2D) 7p14 - Formas recesivas (ver CMT 4) NSMH III [enfermedad de Dejerine-Sottas (EDS)] - EDS A Mutaciones gen PMP-22 (AD, AR) - EDS B Mutaciones gen P0 - EDS - C Mutaciones gen EGR2 (10q) - EDS - D (herencia AD) 8q23-q24 desconocida - EDS - E (herencia AR) Localización genética CMT 4 (herencia AR) - CMT 4A (desmielinización) y bulbos cebolla membranas basales) 8q CMT 4B (desmielinización y plegamientos mielínicos focales) 11q23.1
 - CMT 4A (desmielinización) y bulbos cebolla membranas basales) 8q13-21.")
12 - CMT 4C (forma desmielinizante argelina) 5q CMT 4D (enfermedad de Lom) 8q24 - CMT 4E (con velocidad de desconocida conducción preservada) Localización genética - CMT 4F (como EDS-A) 1q22 desconocida Formas complejas Localización genética 1.2. Neuropatía hereditaria por vulnerabilidad excesiva a la presión (NHVP) NSVP Delecciones gen PMP-22 Mutaciones puntiformes gen PMP-22 Localización genética desconocida 1.3. Polineuropatías amiloidóticas familiares (PAF) PAF relacionada con transretina Mutaciones gen transtirretina (18q11) apolipoproteina 1 Idem con apolipoproteina 1 Mutaciones gen
 NSVP Delecciones gen PMP-22 Mutaciones puntiformes gen PMP-22 Localización")
13 Idem con gelsolina Mutaciones gen gelsolina 1.4. Neuropatías sensitivas y disautonómicas hereditarias (NSH) NSH I (autosómica dominante) 9q22-q23 NSH II (autosómica recesiva) Localización genética desconocida NSH III (síndrome de Riley-Day) 9q31-32 tirosin-kinasa A NSH IV (con anhidrosis) Mutaciones gen receptor NSH V (con pérdida fibras desconocida mielínicas finas) Localización genética 1.5. Neuronopatías motoras hereditarias (atrofia muscular espinal) 1.6. Neuronopatía bulboespinal ligada al cromosoma X 2. Neuropatías cuyo síndrome polineurítico va acompañado de semiología por afectación de otros sistemas (neurológicos o no neurológicos) 2.1. Trastornos del metabolismo lipídico Leucodistrofias (p.ej. metacromática, de células globoides y adrenoleucodistrofia)
 1.6. Neuronopatía bulboespinal ligada al cromosoma X 2.")
14 Deficiencias de lipoproteinas (p.ej., enfermedades de Tangier y Bassen-Kornzweig) Refsum) Enfermedades por acúmulo de ácido fitánico (enf. de Deficiencia de alfa-galactosidasa (enf. de Fabry) Colestenolosis Lipoidosis por acúmulo de esfingomielina 2.2. Porfirias Aguda intermitente Variegata Coproporfiria hereditaria Déficit de Ala deshidrogenasa 2.3. Trastornos de la reparación del ADN Xeroderma pigmentoso Ataxia telangiectasia Síndrome de Cockayne 2.4. Asociadas a citopatías mitocondriales

15 2.5. Asociadas a ataxias hereditarias 2.6. Otras neuropatías hereditarias Plexopatía braquial Neuropatía axonal gigante Neuroacantocitosis Enfermedad de Chediak-Higashi

16 10

17 12
18 11

Grupo de Estudio de Enfermedades Neuromusculares Sociedad Española de Neurología www.sen.es
 1 NEUROPATIAS SENSITIVAS Y AUTONOMICAS HEREDITARIAS (NSAH) Grupo de Estudio de Enfermedades Neuromusculares Sociedad Española de Neurología www.sen.es documento PDF creado para la web el 7 de Julio del
1 NEUROPATIAS SENSITIVAS Y AUTONOMICAS HEREDITARIAS (NSAH) Grupo de Estudio de Enfermedades Neuromusculares Sociedad Española de Neurología www.sen.es documento PDF creado para la web el 7 de Julio del
Neuropatías hereditarias: Evaluación clínica y mecanismos
 Neuropatías hereditarias: Evaluación clínica y mecanismos patogénicos. RESULTADOS DEL REGISTRO MULTICÉNTRICO ESPAÑOL TREAT CMT DRA. MARÍA DEL MAR GARCÍA ROMERO/ DR. SAMUEL I PASCUAL PASCUAL S. NEUROPEDIATRÍA.
Neuropatías hereditarias: Evaluación clínica y mecanismos patogénicos. RESULTADOS DEL REGISTRO MULTICÉNTRICO ESPAÑOL TREAT CMT DRA. MARÍA DEL MAR GARCÍA ROMERO/ DR. SAMUEL I PASCUAL PASCUAL S. NEUROPEDIATRÍA.
Herencia en humanos. El desarrollo de los rasgos tanto normales como anormales de los organismos es el resultado en forma variable del:
 Herencia en humanos El desarrollo de los rasgos tanto normales como anormales de los organismos es el resultado en forma variable del: Genotipo (información genética contenida en el ADN) Ambiente que actúa
Herencia en humanos El desarrollo de los rasgos tanto normales como anormales de los organismos es el resultado en forma variable del: Genotipo (información genética contenida en el ADN) Ambiente que actúa
Introducción General a la Investigación en Enfermedades Raras
 Introducción eneral a la Investigación en Enfermedades Raras Francesc Palau Profesor de Investigación, n, Instituto de Biomedicina de Valencia, CSIC Director Científico, CIBER de Enfermedades Raras (CIBERER)
Introducción eneral a la Investigación en Enfermedades Raras Francesc Palau Profesor de Investigación, n, Instituto de Biomedicina de Valencia, CSIC Director Científico, CIBER de Enfermedades Raras (CIBERER)
D E B I L I D A D D E M I E M B R O S I N F E R I O R E S : E L PA P E L D E L A G E N É T I C A
 D E B I L I D A D D E M I E M B R O S I N F E R I O R E S : E L PA P E L D E L A G E N É T I C A M E L I S S A F O N TA LV O A C O S TA T U T O R E S : R O C Í O J A D R A Q U E F R A N C I S C O G Ó M
D E B I L I D A D D E M I E M B R O S I N F E R I O R E S : E L PA P E L D E L A G E N É T I C A M E L I S S A F O N TA LV O A C O S TA T U T O R E S : R O C Í O J A D R A Q U E F R A N C I S C O G Ó M
TRASTORNOS NEUROMUSCULARES Y DEL MOVIMIENTO EN LA INFANCIA
 TRASTORNOS NEUROMUSCULARES Y DEL MOVIMIENTO EN LA INFANCIA DR. PATRICIO GUERRA NEURÓLOGO INFANTIL Y ADOLESCENTES MAGÍSTER NEUROCIENCIAS ESCUELA DE MEDICINA UNIVERSIDAD SAN SEBASTIÁN SEDE PATAGONIA PUERTO
TRASTORNOS NEUROMUSCULARES Y DEL MOVIMIENTO EN LA INFANCIA DR. PATRICIO GUERRA NEURÓLOGO INFANTIL Y ADOLESCENTES MAGÍSTER NEUROCIENCIAS ESCUELA DE MEDICINA UNIVERSIDAD SAN SEBASTIÁN SEDE PATAGONIA PUERTO
Enfermedad de Charcot-Marie-Tooth: causas, síntomas, tratamientos y más
 Enfermedad de Charcot-Marie-Tooth: causas, síntomas, tratamientos y más Qué es la enfermedad de Charcot-Marie-Tooth? La enfermedad de Charcot-Marie-Tooth (CMT) es un término genérico para una variedad
Enfermedad de Charcot-Marie-Tooth: causas, síntomas, tratamientos y más Qué es la enfermedad de Charcot-Marie-Tooth? La enfermedad de Charcot-Marie-Tooth (CMT) es un término genérico para una variedad
ENFERMEDAD DE CHATCOT-MARIE-TOOTH (CMT) Dra. Teresa Sevilla Mantecón Hospital La Fe de Valencia
 Dra. Teresa Sevilla Mantecón Hospital La Fe de Valencia") ENFERMEDAD DE CHATCOT-MARIE-TOOTH (CMT) Dra. Teresa Sevilla Mantecón Hospital La Fe de Valencia Índice de contenido: Qué es la enfermedad de Charcot-Marie-Tooth? Cuáles son los síntomas de la enfermedad
ENFERMEDAD DE CHATCOT-MARIE-TOOTH (CMT) Dra. Teresa Sevilla Mantecón Hospital La Fe de Valencia Índice de contenido: Qué es la enfermedad de Charcot-Marie-Tooth? Cuáles son los síntomas de la enfermedad
Diagnóstico electrofisiológico. Valor del estudio electromiografico. El estudio electrofisiológico del sistema nervioso periférico (SNP) debe
 debe") Diagnóstico electrofisiológico. Valor del estudio electromiografico. El estudio electrofisiológico del sistema nervioso periférico (SNP) debe considerarse como una prolongación de la exploración neurológica
Diagnóstico electrofisiológico. Valor del estudio electromiografico. El estudio electrofisiológico del sistema nervioso periférico (SNP) debe considerarse como una prolongación de la exploración neurológica
Ataxias. Jorge Luis Palacios Espichán MIR 3 Análisis Clínicos. XX JORNADA de FORMACIÓN INTERHOSPITALARIA del LABORATORIO CLÍNICO
 XX JORNADA de FORMACIÓN INTERHOSPITALARIA del LABORATORIO CLÍNICO LABORATORIO CLÍNICO EN EL DIAGNÓSTICO DE LAS ENFERMEDADES NEUROLÓGICAS Ataxias Jorge Luis Palacios Espichán MIR 3 Análisis Clínicos Ataxia
XX JORNADA de FORMACIÓN INTERHOSPITALARIA del LABORATORIO CLÍNICO LABORATORIO CLÍNICO EN EL DIAGNÓSTICO DE LAS ENFERMEDADES NEUROLÓGICAS Ataxias Jorge Luis Palacios Espichán MIR 3 Análisis Clínicos Ataxia
EVOLUCION NUMERICA DE LAS ENFERMEDADES NEUROGENETICAS
 EVOLUCION NUMERICA DE LAS ENFERMEDADES NEUROGENETICAS AÑO NUMERO 1966 1.500 1994 6.700 72% autosómico dominante 20% autosómico recesivo 6% ligadas al X 0,5% ligadas al Y 1,5 % debidas a genes mitocondriales
EVOLUCION NUMERICA DE LAS ENFERMEDADES NEUROGENETICAS AÑO NUMERO 1966 1.500 1994 6.700 72% autosómico dominante 20% autosómico recesivo 6% ligadas al X 0,5% ligadas al Y 1,5 % debidas a genes mitocondriales
GUÍA SOBRE ATROFIA MUSCULAR ESPINAL
 GUÍA SOBRE ATROFIA MUSCULAR ESPINAL Teleton 05_atrofia mus.indd 1 ÍNDICE 3... Qué es la atrofia muscular espinal? 3... Cómo se adquiere? 3... Cómo se manifiesta? 6... Cómo se realiza el diagnóstico? 7...
GUÍA SOBRE ATROFIA MUSCULAR ESPINAL Teleton 05_atrofia mus.indd 1 ÍNDICE 3... Qué es la atrofia muscular espinal? 3... Cómo se adquiere? 3... Cómo se manifiesta? 6... Cómo se realiza el diagnóstico? 7...
Polineuropatías. Prof. Juan Jiménez Alonso. Curso _asistenciales/medicina_int erna/documentos.
 Polineuropatías Curso 2008-2009 http://www.hvn.es/servicios _asistenciales/medicina_int erna/documentos.php Clasificación Adquiridas inmunitarias : Guillain-Barré (aguda), PN desmielinizante crónica y
Polineuropatías Curso 2008-2009 http://www.hvn.es/servicios _asistenciales/medicina_int erna/documentos.php Clasificación Adquiridas inmunitarias : Guillain-Barré (aguda), PN desmielinizante crónica y
Clonación de genes de enfermedades humanas Curso de Genética Molecular Ciencias Biológicas Universidad de Jaén
 Clonación de genes de enfermedades humanas Curso de Genética Molecular Ciencias Biológicas Universidad de Jaén Antonio Caruz Arcos Dpto. Biología Experimental, Área de Genética Universidad de Jaén Prevalencia
Clonación de genes de enfermedades humanas Curso de Genética Molecular Ciencias Biológicas Universidad de Jaén Antonio Caruz Arcos Dpto. Biología Experimental, Área de Genética Universidad de Jaén Prevalencia
ALGORITMO DIAGNÓSTICO DE LAS NEUROPATÍAS ADQUIRIDAS Julio Pardo Fernández Servicio de Neurología. Hospital Clínico. Santiago de Compostela.
 ALGORITMO DIAGNÓSTICO DE LAS NEUROPATÍAS ADQUIRIDAS Julio Pardo Fernández Servicio de Neurología. Hospital Clínico. Santiago de Compostela. Las polineuropatías constituyen una patología neurológica frecuente
ALGORITMO DIAGNÓSTICO DE LAS NEUROPATÍAS ADQUIRIDAS Julio Pardo Fernández Servicio de Neurología. Hospital Clínico. Santiago de Compostela. Las polineuropatías constituyen una patología neurológica frecuente
TEMA 8 NEUROPATOLOGÍA DEL NERVIO PERIFÉRICO
 TEMA 8 NEUROPATOLOGÍA DEL NERVIO PERIFÉRICO DRA. AURORA ASTUDILLO GLEZ. MD PHD DEPARTAMENTO DE CIRUGÍA Y ESPECIALIDADES MÉDICO QUIRÚRGICAS FACULTAD DE MEDICINA UNIVERSIDAD DE OVIEDO PATOLOGÍA DEL NERVIO
TEMA 8 NEUROPATOLOGÍA DEL NERVIO PERIFÉRICO DRA. AURORA ASTUDILLO GLEZ. MD PHD DEPARTAMENTO DE CIRUGÍA Y ESPECIALIDADES MÉDICO QUIRÚRGICAS FACULTAD DE MEDICINA UNIVERSIDAD DE OVIEDO PATOLOGÍA DEL NERVIO
26 1. FACTORES QUE MODIFICAN LOS PATRONES DE HERENCIA MONOGENICA
 Tema 26 1. FACTORES QUE MODIFICAN LOS PATRONES DE HERENCIA MONOGENICA Heterogeneidad alélica y/o de locus Penetrancia incompleta Expresividad Variable Anticipación Sello genómico Pleiotropía Mutaciones
Tema 26 1. FACTORES QUE MODIFICAN LOS PATRONES DE HERENCIA MONOGENICA Heterogeneidad alélica y/o de locus Penetrancia incompleta Expresividad Variable Anticipación Sello genómico Pleiotropía Mutaciones
Repaso Herencia Mendeliana
 Repaso Herencia Mendeliana Primera ley de Mendel: Los alelos segregan. Los dos miembros de una pareja génica segregan en proporciones 1:1. La mitad de los gametos lleva un miembro de la pareja y la otra
Repaso Herencia Mendeliana Primera ley de Mendel: Los alelos segregan. Los dos miembros de una pareja génica segregan en proporciones 1:1. La mitad de los gametos lleva un miembro de la pareja y la otra
PROYECTO: BASES GENÉTICAS DE LA ENFERMEDAD DE BEHÇET. Norberto Ortego Centeno
 PROYECTO: BASES GENÉTICAS DE LA ENFERMEDAD DE BEHÇET Norberto Ortego Centeno La herencia en las enfermedades Son enfermedades hereditarias las que se transmiten de padres a hijos La información viaja en
PROYECTO: BASES GENÉTICAS DE LA ENFERMEDAD DE BEHÇET Norberto Ortego Centeno La herencia en las enfermedades Son enfermedades hereditarias las que se transmiten de padres a hijos La información viaja en
ENFERMEDADES DE LA MOTONEURONA Y ESCLEROSIS LATERAL AMIOTROFICA
 ENFERMEDADES DE LA MOTONEURONA Y ESCLEROSIS LATERAL AMIOTROFICA Clase de neurología. Dr. Jaume Coll i Cantí UAB. 2011 ELA (ESCLEROSIS LATERAL AMIOTRÓFICA) es la más común de las enfermedades de la neurona
ENFERMEDADES DE LA MOTONEURONA Y ESCLEROSIS LATERAL AMIOTROFICA Clase de neurología. Dr. Jaume Coll i Cantí UAB. 2011 ELA (ESCLEROSIS LATERAL AMIOTRÓFICA) es la más común de las enfermedades de la neurona
POSITIVOS. Hipoestesia postural A-delta frío Frío parestésico Hipoestesia para el frío A-delta nociceptivas
 Tipo de fibra Función Sensitivas: A-beta tacto A-beta posición.vibración A-delta frío A-delta dolor agudo Amielínica C dolor quemante Amielínica C tibieza Motoras Autonómicas TIPO DE FIBRA SINTOMAS POSITIVOS
Tipo de fibra Función Sensitivas: A-beta tacto A-beta posición.vibración A-delta frío A-delta dolor agudo Amielínica C dolor quemante Amielínica C tibieza Motoras Autonómicas TIPO DE FIBRA SINTOMAS POSITIVOS
Diagnóstico Genético Preimplantacional Indicaciones Actuales. Dr. Joaquín Moreno Valencia Febrero 2009
 Diagnóstico Genético Preimplantacional Indicaciones Actuales Dr. Joaquín Moreno Valencia Febrero 2009 DGP El diagnóstico Genético Preimplantacional(DGP) se presenta como una forma muy precoz de diagnóstico
Diagnóstico Genético Preimplantacional Indicaciones Actuales Dr. Joaquín Moreno Valencia Febrero 2009 DGP El diagnóstico Genético Preimplantacional(DGP) se presenta como una forma muy precoz de diagnóstico
Neuropatías por atrapamiento Formas Clínicas i diagnóstico eletromiográfico
 . Neuropatías por atrapamiento Formas Clínicas i diagnóstico eletromiográfico Jaume Coll Cantí 1 Neuropatías focales Frecuencia neuropatias focales (n=2426) 2% 1% 4% 8013 EMG El 31% son N. focales 5% 4%
. Neuropatías por atrapamiento Formas Clínicas i diagnóstico eletromiográfico Jaume Coll Cantí 1 Neuropatías focales Frecuencia neuropatias focales (n=2426) 2% 1% 4% 8013 EMG El 31% son N. focales 5% 4%
o Y Cromosomas autosómicos: 22 pares Cromosoma sexual: 1 par (X o Y)
") Cromosomas autosómicos: 22 pares o Y Cromosoma sexual: 1 par (X o Y) Un alelo es dominante cuando su presencia se manifiesta siempre en el fenotipo, y el fenotipo es igual para el homocigótico que para
Cromosomas autosómicos: 22 pares o Y Cromosoma sexual: 1 par (X o Y) Un alelo es dominante cuando su presencia se manifiesta siempre en el fenotipo, y el fenotipo es igual para el homocigótico que para
Solicitud de estudio genético - Neurología
 Solicitud de estudio genético - Neurología 1 Identificación del paciente y datos de la muestra Paciente Apellidos, nombre Fecha de nacimiento Sexo V M Sangre Sangre periférica entre 3 y 5 ml en tubos EDTA
Solicitud de estudio genético - Neurología 1 Identificación del paciente y datos de la muestra Paciente Apellidos, nombre Fecha de nacimiento Sexo V M Sangre Sangre periférica entre 3 y 5 ml en tubos EDTA
Enfermedades hereditarias monogénicas
 Enfermedades hereditarias monogénicas Las enfermedades monogénicas son aquellas producidas por alteraciones en la secuencia de ADN de un solo gen. Los genes son pequeños segmentos de ADN. Están dispuestos
Enfermedades hereditarias monogénicas Las enfermedades monogénicas son aquellas producidas por alteraciones en la secuencia de ADN de un solo gen. Los genes son pequeños segmentos de ADN. Están dispuestos
ENFERMEDADES NEUROLÓGICAS HEREDITARIAS
 6 Durante los últimos 50 años, especialmente desde el establecimiento del número correcto de cromosomas del cariotipo humano en 46 para las células somáticas, la genética ha ido introduciéndose en el campo
6 Durante los últimos 50 años, especialmente desde el establecimiento del número correcto de cromosomas del cariotipo humano en 46 para las células somáticas, la genética ha ido introduciéndose en el campo
09/11/2010. Grado Medicina. Genética 1 er Curso. Genética clínica y consejo genético
 Grado Medicina Genética 1 er Curso TEMA 10 GENÉTICA CLÍNICA. 10.1 Genética clínica y consejo genético. 10.2 Enfermedades de herencia autosómica. 10.3 Enfermedades de herencia ligada al cromosoma X. 10.4
Grado Medicina Genética 1 er Curso TEMA 10 GENÉTICA CLÍNICA. 10.1 Genética clínica y consejo genético. 10.2 Enfermedades de herencia autosómica. 10.3 Enfermedades de herencia ligada al cromosoma X. 10.4
FENOMENOS QUE DIFICULTAN EL ANALISIS DE LA SEGREGACIÓN MENDELIANA
 FENOMENOS QUE DIFICULTAN EL ANALISIS DE LA SEGREGACIÓN MENDELIANA MSc Dra. María Teresa Lemus Valdés Esp. I y II Grado Genética Clínica Profesora e Investigadora Auxiliar OBJETIVOS EXPLICAR los fenómenos
FENOMENOS QUE DIFICULTAN EL ANALISIS DE LA SEGREGACIÓN MENDELIANA MSc Dra. María Teresa Lemus Valdés Esp. I y II Grado Genética Clínica Profesora e Investigadora Auxiliar OBJETIVOS EXPLICAR los fenómenos
Evaluación neurofisiológica del niño y el adolescente con debilidad muscular
 Evaluación neurofisiológica del niño y el adolescente con debilidad muscular Lidia Cabañes Martínez Servicio de Neurofisiología Clínica Enfermedades Musculares en la Infancia y Adolescencia (XII) 26 y
Evaluación neurofisiológica del niño y el adolescente con debilidad muscular Lidia Cabañes Martínez Servicio de Neurofisiología Clínica Enfermedades Musculares en la Infancia y Adolescencia (XII) 26 y
CÁTEDRA FIVAN 5 de Marzo de 2008
 CÁTEDRA FIVAN 5 de Marzo de 2008 CÁTEDRA FIVAN 5 de Marzo de 2008 El GENOMA HUMANO 1 Genoma 75-100 trillones de células 23 pares de cromosomas por célula 3000 millones de pares de bases 20.000-30.000 genes
CÁTEDRA FIVAN 5 de Marzo de 2008 CÁTEDRA FIVAN 5 de Marzo de 2008 El GENOMA HUMANO 1 Genoma 75-100 trillones de células 23 pares de cromosomas por célula 3000 millones de pares de bases 20.000-30.000 genes
Lactante Hipotónico. Dra. P. López Esteban Neurofisiología Clínica. 22 de marzo de 2012
 Lactante Hipotónico Dra. P. López Esteban Neurofisiología Clínica 22 de marzo de 2012 Es realmente necesaria la electromiografía en el diagnostico de las enfermedades musculares en el momento actual? Origen
Lactante Hipotónico Dra. P. López Esteban Neurofisiología Clínica 22 de marzo de 2012 Es realmente necesaria la electromiografía en el diagnostico de las enfermedades musculares en el momento actual? Origen
ESCLEROSIS MÚLTIPLE. -Enfermedad autoinmune, pertenece al grupo de las enfermedades desmielinizantes.
 CÁTEDRA CLÍNICA APLICADA 4 AÑO ESCLEROSIS MÚLTIPLE -Enfermedad autoinmune, pertenece al grupo de las enfermedades desmielinizantes. -Una de las patologías neurológicas más graves (por su cronicidad, severidad,
CÁTEDRA CLÍNICA APLICADA 4 AÑO ESCLEROSIS MÚLTIPLE -Enfermedad autoinmune, pertenece al grupo de las enfermedades desmielinizantes. -Una de las patologías neurológicas más graves (por su cronicidad, severidad,
DISCAPACIDADES DE ETIOLOGIA GENETICA: PROYECCIONES DE ATENCION INTEGRAL. Prof. Dra. Aracely Lantigua Cruz CUBA 2004
 DISCAPACIDADES DE ETIOLOGIA GENETICA: PROYECCIONES DE ATENCION INTEGRAL Prof. Dra. Aracely Lantigua Cruz CUBA 2004 GENOMA HUMANO 30 000 A 50 000 GENES NUCLEARES Y EL ADN MITOCONDRIAL. MUTACIONES DE ESTOS
DISCAPACIDADES DE ETIOLOGIA GENETICA: PROYECCIONES DE ATENCION INTEGRAL Prof. Dra. Aracely Lantigua Cruz CUBA 2004 GENOMA HUMANO 30 000 A 50 000 GENES NUCLEARES Y EL ADN MITOCONDRIAL. MUTACIONES DE ESTOS
ENFERMEDADES HEREDITARIAS EN ANIMALES que son las enfermedades hereditarias?
 ENFERMEDADES HEREDITARIAS EN ANIMALES que son las enfermedades hereditarias? Las enfermedades hereditarias son aquellas que se transmiten mediante el proceso de la herencia, es decir de los progenitores
ENFERMEDADES HEREDITARIAS EN ANIMALES que son las enfermedades hereditarias? Las enfermedades hereditarias son aquellas que se transmiten mediante el proceso de la herencia, es decir de los progenitores
Padre con enfermedad. 50% con enfermedad
 a. Autosómica dominante: Padre con 50% con Se presenta cuando la copia del gen alterado es dominante sobre el normal y basta una sola copia para que se exprese la. El gen se encuentra en uno de los 22
a. Autosómica dominante: Padre con 50% con Se presenta cuando la copia del gen alterado es dominante sobre el normal y basta una sola copia para que se exprese la. El gen se encuentra en uno de los 22
Consideraciones genéticas en la enfermedad de Fabry. Dra. Mónica López Rodríguez Servicio Medicina Interna
 Consideraciones genéticas en la enfermedad de Fabry Dra. Mónica López Rodríguez Servicio Medicina Interna 1 DEFINICIÓN GENÉTICA La Genética es el estudio de la herencia, el proceso en el cual un padre
Consideraciones genéticas en la enfermedad de Fabry Dra. Mónica López Rodríguez Servicio Medicina Interna 1 DEFINICIÓN GENÉTICA La Genética es el estudio de la herencia, el proceso en el cual un padre
Atrofia muscular espinal. Ana Camacho
 Atrofia muscular espinal no ligada al gen SMN1 Ana Camacho Sección n de Neurología a Infantil Objetivos Qué es la AMS no ligada a SMN1? Clasificación Tipos principales Casos clínicos Conclusiones Atrofia
Atrofia muscular espinal no ligada al gen SMN1 Ana Camacho Sección n de Neurología a Infantil Objetivos Qué es la AMS no ligada a SMN1? Clasificación Tipos principales Casos clínicos Conclusiones Atrofia
Enfermedad de Charcot-Marie-Tooth en numerosos miembros de una familia
 MEDISAN 2016;20(2):210 CASO CLÍNICO Enfermedad de Charcot-Marie-Tooth en numerosos miembros de una familia Charcot-Marie-Tooth disease in several members of a family Dra. Tamara Rubio González, I Al. Lisandra
MEDISAN 2016;20(2):210 CASO CLÍNICO Enfermedad de Charcot-Marie-Tooth en numerosos miembros de una familia Charcot-Marie-Tooth disease in several members of a family Dra. Tamara Rubio González, I Al. Lisandra
EXPLORACION NEUROFISIOLOGICA DEL LACTANTE HIPOTONICO. Dra. P. López Esteban Neurofisiología Clínica
 EXPLORACION NEUROFISIOLOGICA DEL LACTANTE HIPOTONICO Neurofisiología Clínica Es realmente necesaria la electromigrafía en el diagnostico de las enfermedades musculares en el momento actual?? Origen Periférico
EXPLORACION NEUROFISIOLOGICA DEL LACTANTE HIPOTONICO Neurofisiología Clínica Es realmente necesaria la electromigrafía en el diagnostico de las enfermedades musculares en el momento actual?? Origen Periférico
Árboles genealógicos. Ejercicios de genética
 Árboles genealógicos Ejercicios de genética Qué tipos de problemas podemos resolver? Y más importante aún cómo lo hacemos? Para qué sirve? Identificación de características de origen genético en humanos
Árboles genealógicos Ejercicios de genética Qué tipos de problemas podemos resolver? Y más importante aún cómo lo hacemos? Para qué sirve? Identificación de características de origen genético en humanos
Lactante Hipotónico. Dra. P. López Esteban Neurofisiología Clínica. marzo de 2016
 Lactante Hipotónico Dra. P. López Esteban Neurofisiología Clínica marzo de 2016 Es realmente necesaria la electromiografía en el diagnostico de las enfermedades musculares en el momento actual? Origen
Lactante Hipotónico Dra. P. López Esteban Neurofisiología Clínica marzo de 2016 Es realmente necesaria la electromiografía en el diagnostico de las enfermedades musculares en el momento actual? Origen
Hay numerosas formas de neuropatía
 INTRODUCCI N Hay numerosas formas de neuropatía hereditaria (Tabla 1). La clasificación de Dyck ampliamente usada, divide las neuropatías hereditarias en formas sensitivomotora (HMSN) y sensitiva- autonómica
INTRODUCCI N Hay numerosas formas de neuropatía hereditaria (Tabla 1). La clasificación de Dyck ampliamente usada, divide las neuropatías hereditarias en formas sensitivomotora (HMSN) y sensitiva- autonómica
APROXIMACIÓN AL DIAGNÓSTICO DE LOS ERRORES CONGÉNITOS DEL METABOLISMO DE PRESENTACIÓN EN EL ADULTO A PARTIR DE LAS MANIFESTACIONES CLÍNICAS
 APROXIMACIÓN AL DIAGNÓSTICO DE LOS ERRORES CONGÉNITOS DEL METABOLISMO DE PRESENTACIÓN EN EL ADULTO A PARTIR DE LAS MANIFESTACIONES CLÍNICAS Ana María Bielsa Masdeu Servicio de Medicina Interna del Hospital
APROXIMACIÓN AL DIAGNÓSTICO DE LOS ERRORES CONGÉNITOS DEL METABOLISMO DE PRESENTACIÓN EN EL ADULTO A PARTIR DE LAS MANIFESTACIONES CLÍNICAS Ana María Bielsa Masdeu Servicio de Medicina Interna del Hospital
Figura 1. Unidad Motora
 INTERFACE HARDWARE-SOFTWARE PARA LA MEDICIÓN DE FUERZA PRENSIL EN PACIENTES PEDIATRICOS Carlos Acosta a, Raquel Avila a, Alejandro Martinez a a Coordinación Académica Región Altiplano, Matehuala. San Luis
INTERFACE HARDWARE-SOFTWARE PARA LA MEDICIÓN DE FUERZA PRENSIL EN PACIENTES PEDIATRICOS Carlos Acosta a, Raquel Avila a, Alejandro Martinez a a Coordinación Académica Región Altiplano, Matehuala. San Luis
Lactante Hipotónico. Dra. P. López Esteban Neurofisiología Clínica de marzo de 2013
 Lactante Hipotónico Dra. P. López Esteban Neurofisiología Clínica 21-22 de marzo de 2013 Origen Periférico Asta anterior Nervio Unión Neuromuscular Músculo La evaluación clínica del lactante hipotónico
Lactante Hipotónico Dra. P. López Esteban Neurofisiología Clínica 21-22 de marzo de 2013 Origen Periférico Asta anterior Nervio Unión Neuromuscular Músculo La evaluación clínica del lactante hipotónico
1. Introducción. 3. Fallo Ovárico Precoz en el Síndrome X-frágil
 1. Introducción 2. Síndrome X-frágil y el gen FMR1 2.1 Genética y transmisión del Síndrome X-frágil 2.2 Correlaciones entre genotipo y fenotipo 2.3 Mujeres portadoras de Premutación 3. Fallo Ovárico Precoz
1. Introducción 2. Síndrome X-frágil y el gen FMR1 2.1 Genética y transmisión del Síndrome X-frágil 2.2 Correlaciones entre genotipo y fenotipo 2.3 Mujeres portadoras de Premutación 3. Fallo Ovárico Precoz
CASO CLINICO AUTOPSIA 07 A 7. Dra. Juliana Escobar Stein. Servicio Anatomía Patológica Hospital Valle del Nalón
 CASO CLINICO AUTOPSIA 07 A 7 Dra. Juliana Escobar Stein Servicio Anatomía Patológica Hospital Valle del Nalón 2003 HISTORIA CLINICA Ingresa servicio de psiquiatría Mujer 20 años Soltera Natural de Ecuador
CASO CLINICO AUTOPSIA 07 A 7 Dra. Juliana Escobar Stein Servicio Anatomía Patológica Hospital Valle del Nalón 2003 HISTORIA CLINICA Ingresa servicio de psiquiatría Mujer 20 años Soltera Natural de Ecuador
La interacción de factores genéticos y ambientales Evolución y Desarrollo. Aldo Ferreres Neurofisiología Cátedra 1
 La interacción de factores genéticos y ambientales Evolución y Desarrollo Aldo Ferreres Neurofisiología Cátedra 1 Contenidos Factores genéticos y ambientales Fenotipo y genotipo Interacción de factores
La interacción de factores genéticos y ambientales Evolución y Desarrollo Aldo Ferreres Neurofisiología Cátedra 1 Contenidos Factores genéticos y ambientales Fenotipo y genotipo Interacción de factores
DÍA MUNDIAL DEL ALZHEIMER JORNADA 20º ANIVERSARIO DE AFA BIZKAIA
 OSASUN SAILA Osasun Sailburua DEPARTAMENTO DE SANIDAD Consejero de Sanidad DÍA MUNDIAL DEL ALZHEIMER JORNADA 20º ANIVERSARIO DE AFA BIZKAIA PALABRAS DEL CONSEJERO DE SANIDAD Bilbao, 18 de septiembre de
OSASUN SAILA Osasun Sailburua DEPARTAMENTO DE SANIDAD Consejero de Sanidad DÍA MUNDIAL DEL ALZHEIMER JORNADA 20º ANIVERSARIO DE AFA BIZKAIA PALABRAS DEL CONSEJERO DE SANIDAD Bilbao, 18 de septiembre de
Una mujer de 25 anos expresa la hemophilia A de forma leve, la evaluación genética revela
 1. Una mujer de 25 anos expresa la hemophilia A de forma leve, la evaluación genética revela que ella es una portadora heterocigotica para la mutación del gen del facto VIII de los sgtes enunciados explica
1. Una mujer de 25 anos expresa la hemophilia A de forma leve, la evaluación genética revela que ella es una portadora heterocigotica para la mutación del gen del facto VIII de los sgtes enunciados explica
HOSPITAL LA VICTORIA
 OCTUBRE DE 1997 COLECCION DERECHO A VIVIR EN DESVENTAJA FOLLETO No. 4 ASESORAMIENTO GENÉTICO AUTORES: JUAN CARLOS PRIETO RIVERA., MD,MSc (1) OLGA GUTIÉRREZ U. Bact. (1) 1: Hospital la Victoria - Secretaría
OCTUBRE DE 1997 COLECCION DERECHO A VIVIR EN DESVENTAJA FOLLETO No. 4 ASESORAMIENTO GENÉTICO AUTORES: JUAN CARLOS PRIETO RIVERA., MD,MSc (1) OLGA GUTIÉRREZ U. Bact. (1) 1: Hospital la Victoria - Secretaría
CASOS CLÍNICOS PARA DIFERENCIAR EL SISTEMA NERVIOSO CENTRAL DEL PERIFÉRICO
 CASOS CLÍNICOS PARA DIFERENCIAR EL SISTEMA NERVIOSO CENTRAL DEL PERIFÉRICO Daniel Sanchez Masian Neurology/Neurosurgery Unit Animal Health Trust Lanwades Park, Kentford Newmarket, Suffolk CB8 7UU Las enfermedades
CASOS CLÍNICOS PARA DIFERENCIAR EL SISTEMA NERVIOSO CENTRAL DEL PERIFÉRICO Daniel Sanchez Masian Neurology/Neurosurgery Unit Animal Health Trust Lanwades Park, Kentford Newmarket, Suffolk CB8 7UU Las enfermedades
Síndrome de Ambras, Características, Síntomas, Causas, Diagnóstico y Tratamientos
 Síndrome de Ambras, Características, Síntomas, Causas, Diagnóstico y Tratamientos El síndrome de Ambras, también conocido como síndrome del Hombre Lobo o hipertricosis, es una variedad rara de la hipertricosis
Síndrome de Ambras, Características, Síntomas, Causas, Diagnóstico y Tratamientos El síndrome de Ambras, también conocido como síndrome del Hombre Lobo o hipertricosis, es una variedad rara de la hipertricosis
POLINEUROPATIA Y DOLOR NEUROPATICO
 POLINEUROPATIA Y DOLOR NEUROPATICO DR MARIO FUENTEALBA UDEC - HGGB CONTENIDO DIAGNOSTICO DE POLINEUROPATIA ESTUDIO DE POLINEUROPATIA FISIOPATOLOGIA DOLOR NEUROPATICO MANEJO DE DOLOR NEUROPATICO EPIDEMIOLOGIA
POLINEUROPATIA Y DOLOR NEUROPATICO DR MARIO FUENTEALBA UDEC - HGGB CONTENIDO DIAGNOSTICO DE POLINEUROPATIA ESTUDIO DE POLINEUROPATIA FISIOPATOLOGIA DOLOR NEUROPATICO MANEJO DE DOLOR NEUROPATICO EPIDEMIOLOGIA
Importancia de la genética en pediatría
 Neonatología Tema 2 Genética. Clasificación de las anomalías cromosómicas. Estudio de los principales cuadros. Diagnóstico prenatal: indicaciones y metodología Importancia de la genética en pediatría Ambiental
Neonatología Tema 2 Genética. Clasificación de las anomalías cromosómicas. Estudio de los principales cuadros. Diagnóstico prenatal: indicaciones y metodología Importancia de la genética en pediatría Ambiental
COMPLICACIONES NEUROLÓGICAS ASOCIADAS CON LA INFECCIÓN POR EL VIRUS DE LA INMUNODEFICIENCIA HUMANA
 COMPLICACIONES NEUROLÓGICAS ASOCIADAS CON LA INFECCIÓN POR EL VIRUS DE LA INMUNODEFICIENCIA HUMANA Gonzalo Zúñiga M.D. Neurólogo, Profesor de la Facultad de Salud Universidad del Valle, Cali - Colombia.
COMPLICACIONES NEUROLÓGICAS ASOCIADAS CON LA INFECCIÓN POR EL VIRUS DE LA INMUNODEFICIENCIA HUMANA Gonzalo Zúñiga M.D. Neurólogo, Profesor de la Facultad de Salud Universidad del Valle, Cali - Colombia.
Enfermedades Neuromusculares en la Infancia y Adolescencia (IX)
") Enfermedades Neuromusculares en la Infancia y Adolescencia (IX) Hospital Ramón y Cajal Universidad de Alcalá de Henares Madrid, 23 de marzo del 2012 Distrofia Muscular de Cinturas tipo 2-A Eduardo Gutiérrez-Rivas
Enfermedades Neuromusculares en la Infancia y Adolescencia (IX) Hospital Ramón y Cajal Universidad de Alcalá de Henares Madrid, 23 de marzo del 2012 Distrofia Muscular de Cinturas tipo 2-A Eduardo Gutiérrez-Rivas
1 Congreso Argentino de Medicina Interna Pediátrica
 1 Congreso Argentino de Medicina Interna Pediátrica Hospital pediátrico de alta complejidad. 3 nivel de atención. Atiende pacientes de todo el país y países limítrofes. 9 salas de internación con entre
1 Congreso Argentino de Medicina Interna Pediátrica Hospital pediátrico de alta complejidad. 3 nivel de atención. Atiende pacientes de todo el país y países limítrofes. 9 salas de internación con entre
La bradicinesia se define como la lentitud para iniciar y continuar los movimientos, así como dificultad para ajustar la posición corporal.
 1 CIE-10: VI Enfermedades del sistema nervioso G00-G99 Enfermedad de Parkinson G20 GPC Diagnóstico y tratamiento de la enfermedad de Parkinson inicial en el primer nivel de atención Definición La enfermedad
1 CIE-10: VI Enfermedades del sistema nervioso G00-G99 Enfermedad de Parkinson G20 GPC Diagnóstico y tratamiento de la enfermedad de Parkinson inicial en el primer nivel de atención Definición La enfermedad
CUADRO 1A EXTREMIDADES SUPERIORES. Latencia distal ( miliseg ) Media ± SD ± 3.9. > 20 uv Dedo muñeca ± 4.14
 Media ± SD ± 3.9. > 20 uv Dedo muñeca ± 4.14") CUADRO 1A EXTREMIDADES SUPERIORES Datos de conducción nerviosa normal: referencia de laboratorio de electromiografía de la Universidad de la Alabama y Birmingham adaptado al servicio de Neurofisiología
CUADRO 1A EXTREMIDADES SUPERIORES Datos de conducción nerviosa normal: referencia de laboratorio de electromiografía de la Universidad de la Alabama y Birmingham adaptado al servicio de Neurofisiología
ACTAS I CONGRESO NACIONAL EN ATENCIÓN TEMPRANA RETOS EDUCATIVOS, SOCIALES, TECNOLÓGICOS Y DE LA SALUD EN ATENCIÓN TEMPRANA
 ACTAS I CONGRESO NACIONAL EN ATENCIÓN TEMPRANA RETOS EDUCATIVOS, SOCIALES, TECNOLÓGICOS Y DE LA SALUD EN ATENCIÓN TEMPRANA TÍTULO: DIAGNÓSTICO PRECOZ DEL SÍNDROME DE X-FRÁGIL Y SU IMPLICACIÓN EN ATENCIÓN
ACTAS I CONGRESO NACIONAL EN ATENCIÓN TEMPRANA RETOS EDUCATIVOS, SOCIALES, TECNOLÓGICOS Y DE LA SALUD EN ATENCIÓN TEMPRANA TÍTULO: DIAGNÓSTICO PRECOZ DEL SÍNDROME DE X-FRÁGIL Y SU IMPLICACIÓN EN ATENCIÓN
Neuropatología 3. Patología II (PA2026) UNIBE III cuatrimestre 2012
 UNIBE III cuatrimestre 2012") Neuropatología 3 Patología II (PA2026) UNIBE III cuatrimestre 2012 Temas Enfermedades por priones. Enfermedades desmielinizantes, esclerosis múltiple, encefalomielitis diseminada aguda y leucoencefalitis
Neuropatología 3 Patología II (PA2026) UNIBE III cuatrimestre 2012 Temas Enfermedades por priones. Enfermedades desmielinizantes, esclerosis múltiple, encefalomielitis diseminada aguda y leucoencefalitis
Herencia. Br. Angel E. Hernandez C.
 Herencia Br. Angel E. Hernandez C. Genética Ciencia que estudia la herencia y la variabilidad. Estudia: - Los caracteres hereditarios y la forma en que se transmiten de generación a generación. - Las semejanzas
Herencia Br. Angel E. Hernandez C. Genética Ciencia que estudia la herencia y la variabilidad. Estudia: - Los caracteres hereditarios y la forma en que se transmiten de generación a generación. - Las semejanzas
SISTEMA NERVIOSO PERIFERICO GUÍA DE ESTUDIO CATEDRA CLÍNICA MEDICA Y NEUROLOGÍA CARRERA TERAPIA OCUPACIONAL FACULTAD DE PSICOLOGÍA UBA
 SISTEMA NERVIOSO PERIFERICO GUÍA DE ESTUDIO CATEDRA CLÍNICA MEDICA Y NEUROLOGÍA CARRERA TERAPIA OCUPACIONAL FACULTAD DE PSICOLOGÍA UBA Nervios craneales Son 12 pares de nervios que salen de la base del
SISTEMA NERVIOSO PERIFERICO GUÍA DE ESTUDIO CATEDRA CLÍNICA MEDICA Y NEUROLOGÍA CARRERA TERAPIA OCUPACIONAL FACULTAD DE PSICOLOGÍA UBA Nervios craneales Son 12 pares de nervios que salen de la base del
Grupo de Estudio de Enfermedades Neuromusculares Sociedad Española de Neurología www.sen.es
 1 NEUROPATIAS ADQUIRIDAS Grupo de Estudio de Enfermedades Neuromusculares Sociedad Española de Neurología www.sen.es documento PDF creado para la web el 7 de Julio del 2004 2 NEUROPATIAS ADQUIRIDAS Ver
1 NEUROPATIAS ADQUIRIDAS Grupo de Estudio de Enfermedades Neuromusculares Sociedad Española de Neurología www.sen.es documento PDF creado para la web el 7 de Julio del 2004 2 NEUROPATIAS ADQUIRIDAS Ver
Síndrome periódico asociado a la criopirina (CAPS)
") www.printo.it/pediatric-rheumatology/ar/intro Síndrome periódico asociado a la criopirina (CAPS) Versión de 2016 1. QUÉ SON LOS CAPS 1.1 En qué consiste? El síndrome periódico asociado a la criopirina
www.printo.it/pediatric-rheumatology/ar/intro Síndrome periódico asociado a la criopirina (CAPS) Versión de 2016 1. QUÉ SON LOS CAPS 1.1 En qué consiste? El síndrome periódico asociado a la criopirina
TRASTORNOS DE SANGRADOS.
 1 TRASTORNOS DE SANGRADOS. Una persona que sangra no únicamente puede tener como causa de su sangrado algún defecto de la coagulación, sino también de las plaquetas ó de vasos sanguíneos, de modo que lo
1 TRASTORNOS DE SANGRADOS. Una persona que sangra no únicamente puede tener como causa de su sangrado algún defecto de la coagulación, sino también de las plaquetas ó de vasos sanguíneos, de modo que lo
Enfermedades Raras en Asturias,
 Enfermedades Raras en Asturias, 1996-212 Laura Pruneda González Eva García Fernández Mario Margolles Martins Sistema de Información en Enfermedades Raras de Asturias Dirección General de Salud Pública
Enfermedades Raras en Asturias, 1996-212 Laura Pruneda González Eva García Fernández Mario Margolles Martins Sistema de Información en Enfermedades Raras de Asturias Dirección General de Salud Pública
Capítulo 1 Definición
 Capítulo 1 Definición El 1 de diciembre de 1999, se estableció la definición de enfermedad rara para todas aquellas patologías cuya cifra de prevalencia se encuentra por debajo de 5 casos por cada 10.000
Capítulo 1 Definición El 1 de diciembre de 1999, se estableció la definición de enfermedad rara para todas aquellas patologías cuya cifra de prevalencia se encuentra por debajo de 5 casos por cada 10.000
1. QUÉ SON LAS ENFERMEDADES NEUROMUSCULARES?
 UNIDAD 4. DISCAPACIDAD MOTORA LAS ENFERMEDADES NEUROMUSCULARES. CONTENIDO 1.1 QUÉ SON LAS ENFERMEDADES NEUROMUSCULARES?... 1 1.2. Síntomas... 2 1.3. EPIDEMIOLOGÍA... 3 1.4. causas o etiología... 3 a) Enfermedades
UNIDAD 4. DISCAPACIDAD MOTORA LAS ENFERMEDADES NEUROMUSCULARES. CONTENIDO 1.1 QUÉ SON LAS ENFERMEDADES NEUROMUSCULARES?... 1 1.2. Síntomas... 2 1.3. EPIDEMIOLOGÍA... 3 1.4. causas o etiología... 3 a) Enfermedades
Aspectos generales y las distonías
 Aspectos generales y las distonías 1. Qué es la distonía? Se denomina distonía al cuadro caracterizado por la presencia de movimientos involuntarios secundarios a la contracción simultánea y sostenida
Aspectos generales y las distonías 1. Qué es la distonía? Se denomina distonía al cuadro caracterizado por la presencia de movimientos involuntarios secundarios a la contracción simultánea y sostenida
Universidad Autónoma Metropolitana Unidad Iztapalapa. Licenciatura en Biología Experimental. Centro Médico Nacional Siglo XXI IMSS
 Universidad Autónoma Metropolitana Unidad Iztapalapa Licenciatura en Biología Experimental Centro Médico Nacional Siglo XXI IMSS Análisis de la expresión del complejo Sarcoglicano- Sarsocpan y de proteínas
Universidad Autónoma Metropolitana Unidad Iztapalapa Licenciatura en Biología Experimental Centro Médico Nacional Siglo XXI IMSS Análisis de la expresión del complejo Sarcoglicano- Sarsocpan y de proteínas
La ataxia es, en principio, un síntoma, no es una enfermedad específica o un diagnóstico. Ataxia quiere decir torpeza o pérdida de coordinación.
 DEFINICIÓN: La ataxia es, en principio, un síntoma, no es una enfermedad específica o un diagnóstico. Ataxia quiere decir torpeza o pérdida de coordinación. La ataxia puede afectar a los dedos, manos,
DEFINICIÓN: La ataxia es, en principio, un síntoma, no es una enfermedad específica o un diagnóstico. Ataxia quiere decir torpeza o pérdida de coordinación. La ataxia puede afectar a los dedos, manos,
Cómo detectar la hipercolesterolemia familiar? UNIDAD LIPIDOS
 Cómo detectar la hipercolesterolemia familiar? UNIDAD LIPIDOS Enfermedad hereditaria causada por mutaciones en los genes que regulan el aclaramiento del cldl (receptor de LDL). Herencia autosómica dominante
Cómo detectar la hipercolesterolemia familiar? UNIDAD LIPIDOS Enfermedad hereditaria causada por mutaciones en los genes que regulan el aclaramiento del cldl (receptor de LDL). Herencia autosómica dominante
Ataxia Cerebelosa Subaguda en preescolar de 5 anos.
 Ataxia Cerebelosa Subaguda en preescolar de 5 anos. Chevorn Suzette Adams (R2 Pediatría) Tutor: Dr. Francisco Gómez (Sección de Neuropediatria) HGUA, 17 Diciembre 2014 Antecedentes Personales Embarazo
Ataxia Cerebelosa Subaguda en preescolar de 5 anos. Chevorn Suzette Adams (R2 Pediatría) Tutor: Dr. Francisco Gómez (Sección de Neuropediatria) HGUA, 17 Diciembre 2014 Antecedentes Personales Embarazo
Docente: Dra. M. Gloria Pinto Alumno: Juan Pablo Silva Ramo: Fisiopatología Fecha :
 Docente: Dra. M. Gloria Pinto Alumno: Juan Pablo Silva Ramo: Fisiopatología Fecha : 22-06-2011 Las enfermedades neurodegenerativas, son trastornos del sistema nervioso central marcados por una pérdida
Docente: Dra. M. Gloria Pinto Alumno: Juan Pablo Silva Ramo: Fisiopatología Fecha : 22-06-2011 Las enfermedades neurodegenerativas, son trastornos del sistema nervioso central marcados por una pérdida
Síndrome de Guillain-Barré
 Neuropatías inflamatorias: 1 Síndrome de Guillain-Barré Pilar Mazzetti Soler y el grupo de trabajo de síndrome de Guillain Barré del INSTITUTO NACIONAL DE CIENCIAS NEUROLÓGICAS Se autoriza su difusión
Neuropatías inflamatorias: 1 Síndrome de Guillain-Barré Pilar Mazzetti Soler y el grupo de trabajo de síndrome de Guillain Barré del INSTITUTO NACIONAL DE CIENCIAS NEUROLÓGICAS Se autoriza su difusión
Genética y herencia. Profesora: Marcela Saavedra
 Genética y herencia Profesora: Marcela Saavedra Objetivos Identificar mecanismos de herencia no mendeliana Resolver problemas de genética.?? Teoría cromosómica de la herencia Gregory Mendel no conocía
Genética y herencia Profesora: Marcela Saavedra Objetivos Identificar mecanismos de herencia no mendeliana Resolver problemas de genética.?? Teoría cromosómica de la herencia Gregory Mendel no conocía
EJE D: Aplicaciones biomédicas de la biología
 UNIVERSIDAD NACIONAL DE CORDOBA FACULTAD DE ODONTOLOGIA CATEDRA A DE BIOLOGIA CELULAR 2008 EJE D: Aplicaciones biomédicas de la biología Trabajo Práctico Nº 7: Proliferación Celular y reproducción humana.
UNIVERSIDAD NACIONAL DE CORDOBA FACULTAD DE ODONTOLOGIA CATEDRA A DE BIOLOGIA CELULAR 2008 EJE D: Aplicaciones biomédicas de la biología Trabajo Práctico Nº 7: Proliferación Celular y reproducción humana.
Genetica. Estudio de la Herencia
 Genetica Estudio de la Herencia Introduccion Herencia: la transmision de los rasgos y caracteres de los padres a sus hijos. Genetica: rama de la biologia que estudia las leyes de la herencia. Genetica
Genetica Estudio de la Herencia Introduccion Herencia: la transmision de los rasgos y caracteres de los padres a sus hijos. Genetica: rama de la biologia que estudia las leyes de la herencia. Genetica
CASO CLÍNICO 3 Trastorno de la marcha de origen inferior
 CASO CLÍNICO 3 Trastorno de la marcha de origen inferior Dra. Carmen Pablos Hernández Complejo Asistencial Universitario de Salamanca VALENCIA, 14 DE JUNIO DE 2013 55 Congreso Nacional de la Sociedad Española
CASO CLÍNICO 3 Trastorno de la marcha de origen inferior Dra. Carmen Pablos Hernández Complejo Asistencial Universitario de Salamanca VALENCIA, 14 DE JUNIO DE 2013 55 Congreso Nacional de la Sociedad Española
SISTEMA NERVIOSO PERIFERICO
 SISTEMA NERVIOSO PERIFERICO 31 pares de RAICES NERVIOSAS (y sus prolongaciones) 12 pares de NERVIOS CRANEALES 8 C 12D 5 L 5 S Cada raíz nerviosa está constituida por dos raíces: una raíz dorsal o posterior
SISTEMA NERVIOSO PERIFERICO 31 pares de RAICES NERVIOSAS (y sus prolongaciones) 12 pares de NERVIOS CRANEALES 8 C 12D 5 L 5 S Cada raíz nerviosa está constituida por dos raíces: una raíz dorsal o posterior
PLAN DE NIVELACION DE CIENCIAS NATURALES DEL SEGUNDO PERIODO OCTAVO GRADO 2016
 PLAN DE NIVELACION DE CIENCIAS NATURALES DEL SEGUNDO PERIODO OCTAVO GRADO 2016 1. Las siguientes son funciones de la neurona EXCEPTO: A. Conducir la señal eléctrica B. Recibir información externa interna
PLAN DE NIVELACION DE CIENCIAS NATURALES DEL SEGUNDO PERIODO OCTAVO GRADO 2016 1. Las siguientes son funciones de la neurona EXCEPTO: A. Conducir la señal eléctrica B. Recibir información externa interna
Dra Elizabeth Midón. Dra Carolina Scasso
 Dra Elizabeth Midón Dra Carolina Scasso FP: 63 años, SF, diestra, Mercedes, labores, primaria completa AP: HTA en tto con enalapril 20 mg /día, buen control DM tipo 2 dg 2009 en tto con metformina 500
Dra Elizabeth Midón Dra Carolina Scasso FP: 63 años, SF, diestra, Mercedes, labores, primaria completa AP: HTA en tto con enalapril 20 mg /día, buen control DM tipo 2 dg 2009 en tto con metformina 500
Síndrome de Ehlers-Danlos
 Síndrome de Ehlers-Danlos es el nombre por el que se conocen un grupo heterogéneo de enfermedades hereditarias del tejido conectivo, caracterizadas por hiperlaxitud articular, hiperextensibilidad de la
Síndrome de Ehlers-Danlos es el nombre por el que se conocen un grupo heterogéneo de enfermedades hereditarias del tejido conectivo, caracterizadas por hiperlaxitud articular, hiperextensibilidad de la
DISPRAXIA OCULOMOTORA COMO SIGNO PRECOZ DE DIAGNÓSTICO EN LA ATAXIA-TELANGIECTASIA
 DISPRAXIA OCULOMOTORA COMO SIGNO PRECOZ DE DIAGNÓSTICO EN LA ATAXIA-TELANGIECTASIA Eduardo García Soblechero Margarita Rodríguez Benjumea Adrián García Ron Mª Ángeles Delgado Rioja José Sierra Rodríguez
DISPRAXIA OCULOMOTORA COMO SIGNO PRECOZ DE DIAGNÓSTICO EN LA ATAXIA-TELANGIECTASIA Eduardo García Soblechero Margarita Rodríguez Benjumea Adrián García Ron Mª Ángeles Delgado Rioja José Sierra Rodríguez
TRIPLE PATERNIDAD DRA. ALEXIA ALVAREZ LOZANO
 TRIPLE PATERNIDAD DRA. ALEXIA ALVAREZ LOZANO INFERTILIDAD EN MÉXICO Incremento en los problemas relacionados a la fertilidad humana. Múltipes factores relacionados con la genética y la epigenética del
TRIPLE PATERNIDAD DRA. ALEXIA ALVAREZ LOZANO INFERTILIDAD EN MÉXICO Incremento en los problemas relacionados a la fertilidad humana. Múltipes factores relacionados con la genética y la epigenética del
EPIDERMOLISIS BULLOSA: GENÉTICA Y HERENCIA
 EPIDERMOLISIS BULLOSA: GENÉTICA Y HERENCIA 1) INTRODUCCIÓN: La Epidermolisis Bullosa (EB) es una enfermedad hereditaria y crónica, incurable, cuyo rasgo característico es la formación de ampollas a partir
EPIDERMOLISIS BULLOSA: GENÉTICA Y HERENCIA 1) INTRODUCCIÓN: La Epidermolisis Bullosa (EB) es una enfermedad hereditaria y crónica, incurable, cuyo rasgo característico es la formación de ampollas a partir
Neuroanatomía General:
 Neuroanatomía General: El sistema nervioso humano, es sin lugar a duda, el dispositivo más complejo ideado por la naturaleza. No solo controla todos los procesos que ocurren en nuestro cuerpo recibiendo
Neuroanatomía General: El sistema nervioso humano, es sin lugar a duda, el dispositivo más complejo ideado por la naturaleza. No solo controla todos los procesos que ocurren en nuestro cuerpo recibiendo
Las mutaciones que desencadenan este trastorno suceden en los genes MSH2, MLH1, MSH6 y hpms2.
 Síndrome de Lynch El síndrome de Lynch es una condición genética y hereditaria, que incrementa significativamente el riesgo de padecer cáncer de cólon y en menor porcentaje cáncer de óvarios, útero, estómago,
Síndrome de Lynch El síndrome de Lynch es una condición genética y hereditaria, que incrementa significativamente el riesgo de padecer cáncer de cólon y en menor porcentaje cáncer de óvarios, útero, estómago,
Síndrome de Insuficiencia Respiratoria Aguda. Autora: MsC. Dra. María del Carmen Pino González
 Síndrome de Insuficiencia Respiratoria Aguda Autora: MsC. Dra. María del Carmen Pino González INSUFICIENCIA RESPIRATORIA AGUDA Se conoce como síndrome de insuficiencia respiratoria (SIR) al conjunto de
Síndrome de Insuficiencia Respiratoria Aguda Autora: MsC. Dra. María del Carmen Pino González INSUFICIENCIA RESPIRATORIA AGUDA Se conoce como síndrome de insuficiencia respiratoria (SIR) al conjunto de
Estudios genéticos fetales
 Estudios genéticos fetales Álvaro Gorostiaga Hospital Universitario Basurto, Bilbao Universidad del País Vasco, UPV/EHU Sesión clínica. Unidad de Ecografía y Diagnóstico prenatal Enfermedades genéticas
Estudios genéticos fetales Álvaro Gorostiaga Hospital Universitario Basurto, Bilbao Universidad del País Vasco, UPV/EHU Sesión clínica. Unidad de Ecografía y Diagnóstico prenatal Enfermedades genéticas
Hipercolesterolemias Familiares. Pedro Mata Medicina Interna. Fundación Jiménez Díaz Fundación Hipercolesterolemia Familiar
 Hipercolesterolemias Familiares Pedro Mata Medicina Interna. Fundación Jiménez Díaz Fundación Hipercolesterolemia Familiar Hipercolesterolemia Familiar Heterocigota Trastorno monogénico frecuente (1/400-500)
Hipercolesterolemias Familiares Pedro Mata Medicina Interna. Fundación Jiménez Díaz Fundación Hipercolesterolemia Familiar Hipercolesterolemia Familiar Heterocigota Trastorno monogénico frecuente (1/400-500)
Curs de genètica aplicada a Medicina Fetal
 Curs de genètica aplicada a Medicina Fetal MA Sánchez Durán Diciembre 2015 Introducción Detección de defectos congénitos Desarrollo Genética 2 7 7 Infecciones Monogénicos 55 25 Cromosómicos Poligénico
Curs de genètica aplicada a Medicina Fetal MA Sánchez Durán Diciembre 2015 Introducción Detección de defectos congénitos Desarrollo Genética 2 7 7 Infecciones Monogénicos 55 25 Cromosómicos Poligénico
Síndrome de Percherón, diagnóstico mediante TC. A propósito de un caso.
 Síndrome de Percherón, diagnóstico mediante TC. A propósito de un caso. María Cecilia Escoda Ripoll Mónica Mariana Azor Hospital Español de Mendoza. Introducción: Paciente de sexo femenino consulta por
Síndrome de Percherón, diagnóstico mediante TC. A propósito de un caso. María Cecilia Escoda Ripoll Mónica Mariana Azor Hospital Español de Mendoza. Introducción: Paciente de sexo femenino consulta por